CONFERENCE COVERAGE SERIES
Society for Neuroscience Annual Meeting 2005
Washington, DC, U.S.A.
12 – 16 November 2005
CONFERENCE COVERAGE SERIES
Washington, DC, U.S.A.
12 – 16 November 2005
When it comes to human tests of prospective Alzheimer disease treatments, researchers have learned to be humble. They are familiar with having their expectations raised by solid epidemiology, intriguing biological rationales, and animal model data, only to see them dashed in subsequent clinical trials. This problem has bedeviled trials of nonsteroidal anti-inflammatory drugs (NSAIDs), which either failed to show an effect (e.g., rofecoxib and naproxen [see ARF related news story], and nimesulide [see ARF related news story]), were too toxic (diclofenac [Scharf et al., 1999] and indomethacine [Rogers et al., 1993]), or cut short over questions of cardiovascular side effects (see ARF related news story). Before this backdrop, then, the latest NSAID trial seems worth writing home about, as its results were modestly encouraging.
At the 35th Annual Meeting of the Society for Neuroscience, held last week in Washington, D.C., Sandra Black of the University of Toronto presented phase 2 results of a 31-center trial of R-flurbiprofen, conducted in Canada under Black’s supervision and in the UK under Gordon Wilcock at the University of Bristol. This single enantiomer of flurbiprofen emerged as a candidate from research in the laboratories of Greg Cole and Todd Golde, who tried to find compounds that minimize gastrointestinal toxicity while taking advantage of possible γ-secretase modulating and anti-inflammatory effects (see Morihara et al, 2002; Eriksen et al., 2003; Gasparini et al., 2005). Flurbiprofen is the only approved NSAID still in the running for an AD therapy, though the one discussed here is a version trademarked by Myriad Pharmaceuticals.
At the conference, Black reported that the trial had enrolled 207 people with mild and moderate AD and randomized them into a 400 mg twice daily, 800 mg twice daily regimen, or placebo for one year. After that, 86 Canadian participants enrolled in a blinded follow-on study, and Black reported 6-month data from that, too. Most of the participants were also taking cholinesterase inhibitors. Primary endpoints of the trial included performance on the ADAS-cog battery of neuropsychology tests, the activities of daily living scale used by the ADCS, and the clinical dementia rating (CDR) scale.
Patients who had moderate AD at study outset showed no improvement. People with mild AD showed small improvements, but they were not statistically significant overall. On the cognitive tests, people with mild AD on placebo declined, those on 400 BID R-flurbiprofen declined a bit more slowly, and people on the high dose declined more slowly still. Some of the patients in the follow-on group appeared to regain some cognitive function previously lost to disease, but this effect was small and preliminary. On the assessments of daily activities and global function, the improved performance following high-dose treatment did reach statistical significance. This applied particularly to those patients who had the highest levels of drug in their bloodstream. This phase 2 trial did not collect CSF samples to track distribution of the drug or its effect on Aβ levels, but the next trial will, Black said.
The drug was safe and effective enough for the trial sponsor, Myriad Pharmaceuticals, to begin a phase 3 trial of the compound, trademarked as Flurizan, at 130 centers in the U.S. This trial will compare the 800 mg BID dose to placebo for a year and is currently enrolling patients (see Clinical Trials.gov for more information).
This is not to say that there were no side effects in the phase 2 study. There were, and the list includes transient eosinophilia, anemia, blood pressure elevation, and mild rash. The 1,600 per diem dose necessary to see a significant effect is pushing the limit of the tolerable dose established previously, so a positive outcome of this phase 3 trial is not guaranteed. While R-flurbiprofen shows its colors in people, researchers in academia and industry are continuing to search for new analogs that modulate γ-secretase more strongly without affecting any of its other important substrates (see Peretto et al., 2005).—Gabrielle Strobel.
No Available Further Reading
Oligomeric forms of amyloid-β (and also early pretangle forms of tau) captured much of the buzz at the 35th Annual Meeting of the Society for Neuroscience, held last week in Washington, D.C. Despite some labs' preoccupation with oligomers these days, other groups steadfastly maintain that having a head full of plaques is a dangerous thing in its own right. Immunotherapy appears able to remove plaques and is inching its way through a notoriously fickle clinical trials process. β and γ-secretase remain leading targets for small-molecule drugs, yet whether inhibitors for these enzymes will be able to remove mature plaques is an open question. New evidence shows them to be quite entrenched, at least in mice, and new approaches for removing them may need exploring.
A collaborative research team led by Joanna Jankowsky at California Institute of Technology in Pasadena and David Borchelt’s former group at Johns Hopkins School of Medicine in Baltimore, Maryland, addressed this question genetically. (Borchelt recently moved to the University of Florida, Gainesville.) These scientists wanted a fresh approach to measure in an animal model what happens when Aβ production is suppressed once plaques are in place. After all, people most likely already have plaques when they come to therapy. (PET imaging with Pittsburgh Compound B [Mintun, 2005] is confirming that people with even very mild Alzheimer disease have abundant fibrillar amyloid pathology in their frontal and temporal cortex. Presentations on longitudinal human PIB studies at the SfN conference began reporting that MCI patients who have fibrillar amyloid progress to diagnosis. Researchers even noted cases of patients who appeared normal clinically and neuropsychologically when they entered, but had abundant PIB binding and then declined to an AD diagnosis a few years later.)
To ask what closing the Aβ spigot will do in mice modeling this stage, Jankowsky and colleagues developed a new model that overexpresses the Swedish and Indiana mutations of APP driven by the CamKII promoter with the TET-off system. This allows them to end APP expression at will. They did so at 6 months, when the mice had extensive amyloid plaque pathology, and then kept APP silent for 3 months and 6 months, respectively, before analyzing the mice. Jankowsky showed that amyloid pathology indeed did not progress any further. At the same time, however, the existing pathology stayed stubbornly put: Thioflavin S-positive cored plaques, as well as diffuse amyloid, remained unchanged, and biochemical analysis indicated that both insoluble and soluble Aβ levels remained largely stable. Gliosis stayed in place, as did dystrophic neurites. The study appeared online in PLoS Medicine during the conference (see Jankowsky et al., 2005).
Beyond these measures, Jankowski et al. have not, to date, analyzed neurodegeneration in their new mice. Neither did they present data about behavioral or synaptic deficits. This could address the question of how damaging the residual pathology is, and whether plaques and newly generated, soluble forms of Aβ might be toxic in quite separate ways, as a growing number of researchers are beginning to show.
Jankowsky’s data suggesting that mature brain amyloid is quite entrenched contrasts with previous work reporting amyloid clearance by immunotherapy in mice and humans, and lentiviral delivery in mice of siRNA against BACE (Singer et al., 2005). Her results raise the question of how much secretase inhibitors alone can help. Scientists favor secretase inhibitors because they lie upstream of a pathogenic cascade. Some believe that the different pools of amyloid are connected by dynamic equilibria, so that depleting one form will slowly draw down the others, as well, and the brain will eventually clear them. Jankowsky’s data, however, suggest that clearance of existing amyloid will require separate measures beyond secretase inhibition, for example, immunotherapeutic stimulation of glial cells or activation of Aβ-degrading enzymes.
Can Enzymes Safely Degrade Plaques?
The Aβ-degrading enzymes that are known so far generally remove monomeric or oligomeric forms of Aβ, not the tightly aggregated fibrils that make up dense plaques. At the SfN conference, researchers from Washington University, St. Louis, Missouri, introduced a new player that may nibble away at these stable structures. In side-by-side posters, Xiaoyan Hu and Kejie Yin, working with Jin-Moo Lee and other collaborators at Washington University School of Medicine in St. Louis, in essence propose that when astrocytes gear up to remove plaques, they release the potent enzyme matrix metalloproteinase 9 (MMP-9), which might attack plaques in the human AD brain. MMP-9 belongs to a large family of enzymes that remodel tissues by digesting extracellular matrix components; in the brain they have been implicated in plasticity and regeneration. The Washington University researchers picked up earlier work showing that astrocytes participate in clearing Aβ (see Koistinaho et al., 2004). This study had focused on the role of ApoE in this process. Lee’s group then set out to identify the active proteases. Yin and colleagues first noticed that the activated astrocytes that congregate around plaques in APP/PS1 transgenic mouse models expressed MMP-2 and MMP-9. At the SfN conference, they reported that conditioned medium from cultured astrocytes degrades not only synthetic Aβ into characteristic fragments characterized by mass spectrometry, but also fibrillar Aβ made in vitro. When incubated with slices of APP/PS1 transgenic mice, this astrocyte medium reduced the Aβ load later determined by ELISA.
Hu then compared MMP-9 to neprilysin, IDE, or ECE (but not ACE). Of these enzymes, all degraded soluble Aβ, but only MMP-9 degraded fibrillar Aβ. It did so at sites in the C-terminal hydrophobic region of Aβ that are thought to be important for β-pleated sheet formation. Hu’s poster showed electron microscopy images of Aβ fibrils incubated with these enzymes. The fibrils exposed to MMP-9 looked sparser and showed what appeared to be degraded material. Moreover, Hu incubated fresh brain slices of APP/PS1 mice with each of these proteases and, again, only MMP-9 reduced subsequent thioflavin S staining. Antibodies against MMP-9 lit up astrocytes surrounding plaques in the brains of old APP/PS1 and APPsw mice, as well as compact plaques themselves.
It is important to note that even if further work were to show that MMP-9 indeed helps degrade plaques in AD brain, turbo charging this enzyme does not automatically follow as a therapeutic strategy. MMP-9 has been implicated in the pathogenesis of inflammatory, infectious, and cancerous diseases in many organs. In the brain, it has been implicated in brain tumors such as astrocytomas and is part of an inflammatory cascade. Studies on neuroprotection models of stroke and other conditions suggest that, at least in those instances, MMP-9 needs restraining, not further activation. Further research is needed to sort out how best to handle this voracious enzyme.—Gabrielle Strobel.
No Available Further Reading
This year’s 35th annual meeting of the Society for Neuroscience, held last week in Washington, D.C., left a visitor without a lead news story, such as a new gene or a new experimental therapy. But upon closer inspection, the conference did have groups of researchers share a common sense of excitement about trends afoot, and one such trend revolves around the comeback of the microbule-stabilizing protein tau.
Of course, tau was never really gone. Ever since Alois Alzheimer’s original description of neurofibrillary tangles, it has been clear that tau is a central component of AD. But when the discovery of autosomal-dominant mutations in APP and presenilins in the early 1990s gave researchers something to sink their teeth into, the field’s attention turned overwhelmingly toward the amyloid side of the disease. The discovery a few years later of tau mutations that cause frontotemporal dementia did spur renewed interest in this protein. But these tau mutations do not cause AD, and for years the molecular and cellular biology research that usually follows a major genetic discovery occurred on a separate track from work on Aβ. The twain rarely met, and lectures were routinely given on one protein without much mention of the other. The advent of new tau mouse models has begun to change this, however. At the SfN conference, the Aβ peptide and tau—though not plaques and tangles—were increasingly mentioned together in the same breath.
Beyond the mouse research alone, tau is enjoying a general resurgence of interest across technical fronts. Last week’s SfN conference featured 124 presentations involving tau and AD, almost twice the number of the year before and up from 47 in the year 2000. Aficionados of the field will notice that half the names of presenting researchers in this news series are from labs that formerly investigated mostly the amyloid side of AD. Fueling this renewed interest further is a growing consensus around Virginia Lee’s and John Trojanowski’s observation that tau is an important common pathway to a wide range of neurodegenerative diseases that initially present a wholly different picture to the clinician. Besides AD, this includes Parkinson disease, frontotemporal dementias, and the lysosomal storage disease Pick’s. A large proportion of the major paths to age-related neurodegeneration, each with a different beginning, eventually lead through tau, Frank LaFerla and Eckhard Mandelkow jointly emphasized with Lee at an SfN news conference.
Ironically, just as Alois Alzheimer’s groundbreaking observation is approaching its 100th birthday next year, it appears as if, for all its crystallizing insight, it may have kept investigators pursuing the wrong suspect for decades. One early implication of the new wave of tau research is that it’s not primarily the tangles that cause problems in AD, much as they are realizing that on the amyloid side, it’s not primarily the plaques, either. What follows below and in three cross-linked stories is a four-part summary of tau news highlights from the SfN conference. It is not comprehensive. As always, conventioneers are cordially invited to share their own recollections, comments, corrections. See also Part 2, Part 3, and Part 4.
Tau Can’t Swim Upstream
Frank LaFerla, of the University of California, Irvine, strengthened the emerging connection between amyloid-β and tau as part of his laboratory’s ongoing analysis of the triple transgenic mice expressing APP, PS-1, and tau mutations. A series of 10 presentations at the meeting addressed various aspects involving tau. The scientists’ first major conclusion was that Aβ acts upstream of tau. “When we manipulate Aβ, either genetically or pharmacologically, it has a direct impact on tau. When we augment tau, either genetically or pharmacologically, that has no effect on Aβ. We see a one-way arrow going from Aβ to tau. The arrow going back is absent or small,” LaFerla summarized at a press conference. The body of research suggesting that Aβ acts on tau further downstream in the pathogenic cascade is broadening. Other labs have reported similar evidence, and at the conference Italian researchers from the universities of Florence and Brescia showed data suggesting that the TgCRND8 mutant APP-transgenic mice made in David Westaway’s laboratory (Chishti et al., 2001) develop tau hyperphosphorylation by 7 months of age.
In one presentation by the LaFerla lab, Salvatore Oddo and Antonella Caccamo turned around their previous observation that removing Aβ reduced the tau burden in the triple transgenic mice and asked whether changes in tau could likewise influence the Aβ burden. Before addressing this question directly, Oddo first documented the time course of tau pathology and tau kinase activity in the triple transgenic mice. A panel of p-tau antibodies and Gallyas silver stain on brain sections indicated that tau phosphorylation appeared around 12 months and increased with age. Tau extractions then showed that phosphorylation at the AT100 site precedes tau aggregation, beginning as early as 6 months of age, but phosphorylation at the PHF-1 site sets in only after tau has begun aggregating. Among tau kinases, GSK3β activity showed an increase around 15 months of age; CDK5 levels did not change as the mice aged but the concentration of its activator p25 increased by 15 months, Oddo reported. Perhaps the most important finding from this study came from crossing the triple transgenic mice to another strain that overexpresses human tau sixfold relative to endogenous tau. These tau-laden crosses predictably had increased steady-state levels of tau and more tau phosphorylation, but all this did not change their amyloid pathology at all. Oddo et al. saw no differences in APP or C99 levels, Aβ40 or 42 levels, or amyloid deposits. This expands previous data by this group showing that injection of anti-tau antibodies did not affect amyloid pathology.
Another presentation from the LaFerla group brought up the Aβ-tau connection from a different angle. Kim Green reported that glucocorticoids—stress hormones that are thought to mediate part of the environmental component of a person’s disease risk—increased APP production, BACE activity, intraneuronal Aβ levels, and also hippocampal tau accumulation in the triple transgenic mice. However, glucocorticoid treatment did not increase tau accumulation in transgenic mice lacking APP, suggesting that this effect of stress hormones is a downstream consequence of its effect on APP. (It also implies that glucocorticoid drugs should be prescribed to the elderly with caution.)
LaFerla’s second major conclusion is that soluble forms of tau act importantly in pathogenesis before either tangles or plaques are fully in place, LaFerla noted. This finding agrees with data from two other tau mouse models (Andorfer et al., 2005; SantaCruz et al., 2005). These models differ in that one expresses wild-type human tau without mouse tau and the other expresses an on-off regulatable form of human mutant tau, and yet they both suggest that soluble tau, not the tangles, appears to do the early damage to cognition.
Swim to Platform: Without Tau, Aβ-Loaded Mice Do Fine
Genetically increasing tau may not feed back into amyloid pathology, but what happens in the opposite experiment, that is, decreasing tau in the face of rapid amyloid production? Lennart Mucke of the University of California, San Francisco, chose this approach to address how tau contributes to AD. The cell biology of tau is well-studied and suggests a number of candidate mechanims—Is it the tangles? Loss of microtubule stabilization? Axonal transport? Or the cell cycle?—but in vivo data are more scarce. To address this issue, Erik Roberson in Mucke’s lab bred an APP-transgenic mouse that has well-characterized cognitive deficits and amyloid pathology to a heterogyzous tau deletion mouse. Surprisingly, knocking tau levels in half strongly protected the offspring. The mice with rampant APP expression but little tau performed almost normally in the water maze. They escaped other behavioral abnormalities seen in the APP transgenics. Measures of synaptic function, such as calbindin levels, were almost normal in the tau-depleted mice. By contrast, reducing tau did not change the mice’s amyloid pathology, the presence of dystrophic neurites, or aberrant axonal sprouting, much like increasing tau did not change amyloid pathology in Oddo and LaFerla’s experiment.
This experiment suggests yet again that cognitive performance in mouse models is separate from plaque pathology. Yet the most surprising conclusion from this work may be that the presence of tau—but not of tangles—is necessary for soluble forms of Aβ to wreak the previously noted synaptic and behavioral damage. The implication would be that soluble tau acts downstream of soluble Aβ in a pathway that attacks the function of synapses and, by extension, impairs learning and memory. Carrying this scenario over to humans, this process would happen either before neuritic plaques and neurofibrillary tangles are laid down, or simultaneously in a parallel process. In any event, it would occur outside our ability to measure it at this time. On separate occasions, both Mucke and LaFerla noted that their data call for treating, that is, inhibiting, both tau and Aβ accumulation as a therapeutic strategy.
The tau-transgenic mouse front had more news to offer in Washington. For example, the regulatable r4510Tg tau mouse studied by Karen Ashe’s and Brad Hyman’s labs is reported to develop tangles, neuronal loss, and a memory impairment that improves when soluble tau disappears even as tangles remain stable (SantaCruz et al., 2005). At the conference, these groups presented follow-up data suggesting a dissociation between those neurons that get tangles and those that die. In one study, the scientists used PHF1-staining of tau aggregates and cresyl violet staining of neurons for stereological counts in various subareas of hippocampus, striatum, and cortex of the r4510Tg tau mice to assess tau aggregation in relation to age and neuron loss. They describe varying proportions of hyperphosphorylated tau accumulation and cell death with age in these regions, but the upshot was that tau accumulation did not predict the pattern of neuron loss.
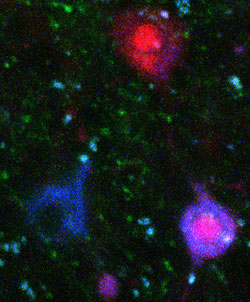
Tangles and Caspases
Multiphoton microscopy shows neurofibrillary tangles (blue) and caspases (red) in the brain of a live tau-transgenic mouse. [Photo courtesy of Tara Spires and Brad Hyman.]
Further, researchers in Hyman's lab have developed a fluorescent reporter that sticks to tangles and are using it with multiphoton microscopy to observe the natural history of tangle growth in live r4510Tg tau mice. First author Tara Spires also reported a way of monitoring caspase activation in vivo. Using RT-PCR, the authors noted an increase of caspase 8 expression in the brains of these mice. Of all the neurons the scientists found to have activated caspase 8, a majority did not have tangles. Taken together, these data hint at a caspase-dependent mechanism of cell death in connection with soluble tau in these mice.
If it’s not the tangles, what, then, is it about the biochemistry of tau that could disturb the function of neurons? Martin Ramsden in Ashe’s lab addresses this question by trying to isolate and characterize a 64 kDa, sarkosyl- insoluble form of tau the scientists hold responsible for the memory deficits in r4510Tg tau mice. Formal characterization of this strain was published this month (Ramsden et al., 2005).—Gabrielle Strobel.
See also Part 2, Part 3, and Part 4 of this series.
 Paul Coleman
Paul Coleman
A host of papers now indicates that neurofibrillary tangles (NFTs) are not a major cause of neuron death. These include an earlier paper by Brad Hyman as well as a paper by Renee Morsch, Bill Simon and me that concluded that neurons live for decades after they have formed NFTs. Although NFTs may not be a major cause of neuron death, they certainly contribute to decreased functional capacity of single neurons as well as synaptic deficits. …More
View all comments by Paul Coleman Cheng-Xin Gong
This is a well-described, interesting study. Together with studies mentioned above by P. Coleman, it is quite likely that neurofibrillary tangles (NFTs) of paired helical filaments (PHFs) themselves do not cause neuronal death in the diseased brain. Instead, formation of highly polymerized NFTs from the unpolymerized, abnormally hyperphosphorylated tau may be a defense mechanism of the affected neurons by which they turn the cytotoxic soluble hyperphosphorylated tau into inert NTFs (Alonso and Iqbal, 2005; Gong et al., 2006).…More
Several studies have demonstrated that unpolymerized hyperphosphorylated tau is neurotoxic, probably due to its sequestering normal tau and other microtubule-associated proteins and, thus, disrupting microtubules (Alonso et al., 1994; 1996; 1997; 2001; Fath et al., 2002), whereas upon polymerization into PHFs/NFTs, tau loses this toxic activity and becomes biologically and pathologically inert (Alonso et al., 2006). This may also explain why some NFT-containing neurons can survive for many years to decades (Morsch et al., 1999).
We have to distinguish the role between abnormally hyperphosphorylated tau and NFTs in the pathogenesis of tauopathies. This and other studies exclude a major role of NFTs in neuronal loss, but do not exclude such a role of the abnormally hyperphosphorylated tau. Actually, the hyperphosphorylated tau is toxic not only in vitro (Alonso et al., 1994; 1996; 1997; 2001), but also in vivo (Wittmann et al., 2001; Jackson et al., 2002). Hyperphosphorylation of tau at different phosphorylation sites has distinct effects on its loss of normal biological activity, its gain of toxic activity, and promoting tau’s self-assembly into PHFs. For example, phosphorylation sites at the middle part of tau (Ser199/Ser202/Thr205, Thr212, Thr231/Ser235, Ser262/Ser356) are critical to convert normal tau to a toxic molecule, whereas phosphorylation at the C-terminus of tau, including the PHF-1 sites (Ser396/Ser404), mainly promotes tau self-assembly into filaments (Abraha et al., 2000; Ferrari et al., 2003; Alonso et al., 2004; Haase et al., 2004). Because accumulated tau, both in human tauopathies and in all animal models of tauopathy, is always hyperphosphorylated, it is very possible that the neurodegeneration seen in these transgenic mouse brains results from unpolymerized hyperphosphorylated tau, whereas polymerization of this toxic tau into inert PHFs/NFTs is the neurons’ self-defense mechanism for survival.
Therefore, this study does not exclude a role of abnormal hyperphosphorylation of tau in neurodegeneration. Future studies on the correlation between time- and region-specific neurodegeneration and the level of unpolymerized hyperphosphorylation of tau, especially at those sites critical to toxicity, in this or transgenic mice with tauopathies, will no doubt provide new insight into the mechanism of tau-mediated neurodegeneration.
Abraha A, Ghoshal N, Gamblin TC, Cryns V, Berry RW, Kuret J, Binder LI. C-terminal inhibition of tau assembly in vitro and in Alzheimer's disease. J Cell Sci. 2000 Nov;113 Pt 21:3737-45. PubMed.
del C Alonso A, Iqbal K. Tau-induced neurodegeneration: a clue to its mechanism. J Alzheimers Dis. 2005 Dec;8(3):223-6. PubMed.
Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001 Jun 5;98(12):6923-8. Epub 2001 May 29 PubMed.
Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996 Jul;2(7):783-7. PubMed.
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994 Jun 7;91(12):5562-6. PubMed.
Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A. 1997 Jan 7;94(1):298-303. PubMed.
Alonso Ad, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A. 2006 Jun 6;103(23):8864-9. PubMed.
Alonso Ad, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004 Aug 13;279(33):34873-81. Epub 2004 Jun 9 PubMed.
Fath T, Eidenmüller J, Brandt R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J Neurosci. 2002 Nov 15;22(22):9733-41. PubMed.
Ferrari A, Hoerndli F, Baechi T, Nitsch RM, Götz J. beta-Amyloid induces paired helical filament-like tau filaments in tissue culture. J Biol Chem. 2003 Oct 10;278(41):40162-8. PubMed.
Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation. J Alzheimers Dis. 2006 Mar;9(1):1-12. PubMed.
Haase C, Stieler JT, Arendt T, Holzer M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J Neurochem. 2004 Mar;88(6):1509-20. PubMed.
Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002 May 16;34(4):509-19. PubMed.
Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999 Feb;58(2):188-97. PubMed.
Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001 Jul 27;293(5530):711-4. Epub 2001 Jun 14 PubMed.
View all comments by Cheng-Xin GongNo Available Further Reading
This is the second installment of a four-part news series about the role of the microtubule-associated protein tau from the 35th Annual Conference of the Society for Neuroscience, held November 12 to 16 in Washington, D.C. See also Introduction and Part 1, Part 3, and Part 4.
Four-Repeat Tau: A Shifty Character
In one of five side-by-side posters showcasing careful expression profile analyses of single neurons from human neurodegenerative disease tissue, Stephen Ginsberg, from the Nathan Kline Institute in Orangeburg, New York, presented new research on tau. Other groups had found that the human brain creates six different isoforms of tau by alternatively splicing the freshly transcribed tau pre-mRNA. Three isoforms come with three tandem repeats, that is, three microtubule binding domains (3R tau), and three other isoforms contain four tandem repeats, that is, four microtubule binding domains (4R tau). In normal brain, 3R and 4R tau occur in about equal measure. Tau mutations in some inherited tauopathies change this ratio prior to tangle formation and cell death (for reviews, see Goedert, 2005; Lee et al., 2001).
Since cholinergic basal forebrain neurons are particularly vulnerable to neurofibrillary tangles in early AD, Ginsberg aspirated single neurons of this type from postmortem tissue of people who at death had been either cognitively normal, diagnosed with mild cognitive impairment, or with AD. Ginsberg collaborated with Elliott Mufson at Rush University Medical Center in Chicago on this study, which used tissue samples from nuns participating in the Religious Orders Study. After having isolated and amplified the mRNA in these single neurons, the scientists used custom-made cDNA arrays to analyze the levels of 3R and 4R tau from these cells. Then they compared the new data to archived data gathered from hippocampal CA1 neurons of a similar grouping of cases, and to data from schizophrenia samples for an additional control. They confirmed earlier knowledge when they found that overall levels of tau expression did not differ among the groups. They hit something new with their finding that people with AD and already with MCI, but not normal aging or schizophrenia, had a shift in the ratio of 3Rtau to 4Rtau such that they had more 4R tau in these particular neurons in basal forebrain and the hippocampal CA1 field. This indicates that not the expression level per se, but some change related to the splicing machinery or regulation may help explain the onset of tauopathies and part of the progression to AD, Ginsberg speculated. This data is in press at the Journal of Neurochemistry.
Speaking more broadly, Ginsberg emphasized the importance of single cell gene expression profiling in order to understand what goes wrong in particular neurons. The regional vulnerability of specific types of neurons remains a mystery in many neurodegenerative diseases. Profiling expression patterns in brain regions, even fairly narrowly circumcised ones, tends to cancel out important effects at the single-cell level, Ginsberg said. “These are complex disorders, where regional vulnerabilities are important. The answer will be in the details, and we need to look at local changes,” Ginsberg said.
Recent technological improvements in laser capture microdissection, RNA isolation and amplification, as well as the design of microarrays and qPCR have led to a wider acceptance of expression profiling from pure populations of cells or from minute quantities of tissue from specific subregions. The SfN conference featured a growing number of presentations in this area. At the same time, researchers are still wrestling with problems such as the minute size of the samples, low yields, and poor integrity of the RNA. Independent validation of the obtained profiles remains a challenge, as well (see Galvin and Ginsberg, 2005).
Toward an RNA-based Tau Treatment
What is it about the tau pre-mRNA that determines the 3R-to-4R shift in tau splicing? And could small-molecule drugs conceivably tweak this process therapeutically? These questions prompted Michael Wolfe’s lab, formerly known as having research interests squarely in the amyloid side of AD research, to enter the tau field. Christine Donahue, a postdoctoral fellow in Wolfe’s lab at Brigham and Women’s Hospital in Boston, picked up a clue from Mike Hutton, who had suggested that the tau pre-mRNA forms a stem loop structure at an exon-intron junction of exon 10. This stem loop, so the idea goes, controls access of a particular splicing factor to its binding site and thus influences whether exon 10 ends up included in the mRNA (this leads to 4R tau) or left out (this yields 3R tau). It was also known that FTDP-17 mutations lead to a decrease in the expression of 3R tau, possibly due to a predicted decrease in stem loop stability.
Donahue approached this question first by stabilizing the proposed tau stem loop in wild-type and FTDP-17 tau with a series of mutations in vitro. This reduced exon 10 inclusion in the splice product and argues for the existence of a stem loop in the tau pre-mRNA. A dementia-causing FTDP-17 point mutation destabilized the stem loop and increased exon 10 inclusion. Next, Donahue added a luciferase reporter to an existing wild-type tau minigene, which allowed her to detect small molecules that stabilize the stem loop. She found that Geneticin, an aminoglycoside antibiotic known to bind to RNA, indeed stabilizes the stem loop and reduces 4R tau production in vitro. Geneticin is neither sufficiently selective nor able to cross the blood-brain barrier (BBB) to become an AD drug itself. However, it proves the principle that tinkering with this RNA structure can influence tau production, and has encouraged an ongoing screen in Wolfe’s lab for more suitable compounds with the same function, Donahue said.
No tau-specific treatments are yet on the horizon, though approaches are entering the pipeline. The drug taxol has shown some promise in animal models, and a search for less toxic microtubule-stabilizing compounds that cross the BBB is underway at the University of Pennsylvania School of Medicine in Philadelphia, Lee said. Illana Gozes, of the Sackler School of Medicine in Tel Aviv, has developed a neuroprotective peptide called NAP (see ARF related news story), which appears to act in a similar way and competes with taxol for binding on microtubules. In Washington, D.C., Gozes reported that a small, double-blind, randomized phase 1a trial of an intranasal formulation of this compound has been completed. Inhibitors for certain tau kinases, for example, GSK3β, are under active study in the pharmaceutical industry. Lithium, an established drug widely used to treat bipolar disorder, also appears to stem tau pathology, and the Alzheimer Disease Cooperative Study is preparing to test it in a clinical trial of patients with mild cognitive impairment starting next year, Lee added. Compared to these approaches, stem loop stabilizers targeting RNA would represent a new generation of mechanism-based drugs; they are at the early discovery stage.—Gabrielle Strobel.
See also Introduction and Part 1, Part 3, and Part 4 of this series.
No Available Comments
No Available Further Reading
This is the third installment of a four-part news series about the role of the microtubule-associated protein tau from the 35th Annual Conference of the Society for Neuroscience, held November 12 to 16 in Washington, D.C. See also Introduction and Part 1, Part 2, and Part 4.
On the Chopping Block
What with Aβ-degrading enzymes up to almost a half-dozen candidates by now (neprilysin, IDE, ECE, ACE, MMP-9; see ARF related SFN story), one would think that an equally impressive phalanx of enzymes is at hand to slice up tau. Alas, tau degradation remains shrouded in mystery. Activated calpain is known to cleave tau in pathological situations, and some researchers have proposed that tau cleavage by caspases promotes neuropathology. The ubiquitin-proteasome pathway has been implicated in pathological situations (see Yang presentation below), but whether it disposes of tau as a normal, or neuroprotective measure remains questionable (David et al., 2002; Feuillette et al., 2005). At the SfN conference, a little-noticed talk at the very end of an afternoon session, when most of the audience had already slipped away, began to fill this gap with a well-substantiated story about an obscure enzyme that relentlessly chops away at tau from the protein’s amino terminal until it is completely degraded.
George Jackson, of the University of California, Los Angeles, presented some of the data gathered in a three-way collaboration among the labs of Daniel Geschwind, also of UCLA; Jackson; and Skip Binder at Northwestern University in Chicago. The story began in Geschwind’s lab, where scientists looked for gene expression differences between cerebellum and cortex of the frontotemporal dementia mouse model P301L. They acted on the hunch that the cerebellum resists tau pathology and neurodegeneration by means of some sort of protective gene expression. This microarray analysis found not only expected genes, such as the apoptosis break bcl2 and the neuroprotective VEGF, but also unexpected ones. This group included PSA, which stands for puromycin-sensitive aminopeptidase. Aminopeptidases are enzymes known to be able to generate free amino acids, but are poorly studied in the human brain. The scientists validated PSA upregulation with Northern blots and in situ hybridization, as well as tissue microarray analysis of normal human and FTD brain where, again, PSA expression was decreased relative to normal brain in vulnerable areas, such as frontal cortex, but increased in resistant areas, such as cerebellum.
Jackson presented in more detail his work validating the function of PSA in a fly model of neurodegeneration. In brief, flies expressing 4R tau (chosen because FTDP mutations lead to a relative increase in 4R tau) in the 800 ommatidia of their compound eyes show a “rough eye” phenotype that allows for ready detection of neurodegeneration. Cross-sections of affected compound eyes reveal a morphology in disarray and cell loss. Genetic tinkering changed that phenotype in ways that jibed with the idea of PSA being a neuroprotective modulator of tau, Jackson reported. That is, coexpressing an inactive mutant form of PSA worsened the 4R tau phenotype, whereas overexpressing active PSA remedied it. PSA suppresses both mutant and wild-type tau phenotypes in flies, and it leads to a decline in tau levels. Binder’s lab then cloned and purified recombinant human tau and incubated it with PSA. The peptidase degraded tau, chewing it up without a trace, and even appeared to show a relative preference for mutant tau over wild-type tau. Puromycin, as well as the peptidase inhibitor bestatin, counteracted this degradation.
This work suggests that certain brain areas upregulate PSA in the presence of mutant tau, and that the enzyme then takes tau apart one amino acid at a time. It is known that PSA expression tends to decline with age, Jackson noted, adding that PSA may make a new therapeutic target. The researchers don’t yet know why PSA appears to be relatively specific for tau, or why it degrades FTDP-mutant tau more readily than wild-type. Interestingly, the PSA gene resides on chromosome 17, near the FTD locus.
Tau Proteomics for a Future Biomarker
The PSA story started out with a gene expression difference, but for other questions it’s necessary to zero in on post-translational changes, which DNA microarrays do not capture. A case in point is the issue of how a slew of changes to existing tau protein occur in relation to disease. A large literature on hyperphosphorylation of tau paired helical filaments, and a smaller one on tau ubiquitination, have made clear that these changes occur and are important. Yet their precise place in the pathogenic cascade has not been determined. It is not clear, for example, when these changes happen relative to the appearance of degradation-resistant forms of tau. One limitation has been that most studies look at some phosphorylation sites but not others, making it difficult to organize and compare existing knowledge quantitatively. Austin Yang at the University of Southern California took a proteomic stab at measuring tau modifications in human tissue more comprehensively. Yang’s team took autopsy samples from early AD brain provided by his collaborator Peter Davies at Albert Einstein College of Medicine in the Bronx, New York. Then the scientists immunopurified soluble pre-tangle tau, digested it with proteases, and ran the peptides through liquid chromatography and high-throughput mass spectrometry. This procedure yielded a complete fingerprint of all tau phosphorylation sites, as well as three amino acids on tau where ubiquitin attaches and elongates poly-ubiquitin chains, Yang said. The data indicate that the formation of ubiquitin chains depends on the phosphorylation state of soluble tau, and that the ubiquitin-proteasome plays a role in the subsequent step of forming protease-resistant tangles. On this issue, Frank LaFerla’s group at the University of California, Irvine, also reported at the conference that blocking the proteasome intensified tau pathology, though these scientists propose that an intraneuronal buildup of Aβ oligomers (see upcoming SfN news summaries) inhibits the proteasome.
Yang’s kind of proteomics analysis is at an early stage, only now moving into exploratory tests with cerebrospinal fluid. However, it is a much richer indicator of pathological tau modifications than the CSF phospho-tau antibody tests currently under development as a diagnostic marker, and could one day replace them, Yang hopes.—Gabrielle Strobel.
See also Introduction and Part 1, Part 2, and Part 4 of this series.
No Available Comments
No Available Further Reading
This is the last installment of a four-part news series about the role of the microtubule-associated protein tau from the 35th Annual Conference of the Society for Neuroscience, held November 12 to 16 in Washington, D.C. See also Introduction and Part 1, Part 2, and Part 3.
Tau and Oxidative Stress
Mouse models also have given researchers new tools to study the relationship between tau and the oxidative stress that results when dysfunctional mitochondria produce too many reactive oxygen species. The conference saw some fruits of this work. Mitochondrial dysfunction has become a point of convergence among many age-related neurodegenerative diseases, and researchers generally assume it to be one of the environmental risk factors that can spur the expression of the neurodegenerative disease for which a person is already at risk. Yet it is still unclear whether it is a primary or secondary event and how it intersects with the disease-specific pathogenic pathways.
In one talk, Ashley Bush of the Mental Health Research Institute, Victoria, Australia, and Massachusetts General Hospital in Charlestown, collaborating with Simon Melov’s lab at the Buck Institute in Novato, California, took advantage of an unusual mouse model to address the question. He used superoxide dismutase 2 (SOD2) knockout mice, whose lack of this radical-scavenging enzyme kills them soon after birth as the radical load overwhelms their peripheral tissues. By treating these mice with catalytic antioxidants, the scientists kept them alive long enough to be able to unmask their brain pathology. When studying their brains at 3 weeks of age, the scientists discovered massive increases in phosphorylation on three tau phosphorylation sites in AD-relevant areas. Increased doses of the antioxidant treatment ameliorated this effect. When the researchers halved the amount of genetically available SOD2 by crossing Tg2576 APP transgenics with heterozygous SOD2 knockouts (who survive and can reproduce), they saw the amyloid burden shoot up, as well. The increase in amyloid was blocked by the antioxidant they administered. The mice also showed changes in metal levels. The lab is now pursuing the relationship among iron, zinc, and phosphorylated tau in more detail.
The data available so far suggest that oxidative stress is a common upstream modulator of both amyloid and tau pathology, Bush noted. For their part, researchers led by Jada Lewis at the Mayo Clinic, Jacksonville, Florida, looked at markers of oxidative stress, such as heme oxygenase-1 and other genes induced in response to it, in their mice expressing the human P301L tau mutant that leads to FTDP-17.
Such work strengthens the case for antioxidative therapies and should lead to better treatments in this area. Many people currently take antioxidant supplements such as vitamin E, beta-carotine, or Ginkgo biloba (Christen, 2004) based on epidemiologic and other data. However, controlled trials of these compounds have mostly been disappointing so far, (e.g., Petersen et al., 2005). Delivering effective doses to the brain has been a major challenge.
Finally, a fresh approach to testing such compounds is worth including in this context, even though it does not offer data on tau yet. Monica Garcia-Alloza in Hyman’s group presented data from her study using the oxidation-sensitive reporter dye Amplex Red in multiphoton microscopy of APP transgenic mice. Working with Brian Bacskai, she found that applying vitamin E or Ginkgo biloba extract (though not beta-carotene) directly to the brain of Tg2576 mice, as well as feeding it orally for 15 days, reduced oxidative stress around plaques. Along with this reduction, she noticed that some curved dendrites were straightening out, similar to what this group had previously described for other experimental therapeutic interventions (Brendza et al., 2005). This suggests that antioxidant therapy might be useful for the structural damage seen with Alzheimer pathology.
The SfN conference had more to offer on tau. The Alzforum will be pleased to post readers' observations and additions as comments to this series.—Gabrielle Strobel.
No Available Further Reading
The story of intraneuronal Aβ, for years that of a few researchers calling into the wind, has generated a steady buzz at the 35th Annual Meeting of the Society for Neuroscience, held November 12-16 in Washington, D.C. Until recently, most researchers have tended to argue that the Aβ peptide can’t be of much consequence inside the neuron, chiefly because neurons secrete it soon after making it. But now, a growing number of laboratories are studying intraneuronal Aβ, and at the conference the topic drew frequent questions after talks and at posters. Active research these days includes the study of intraneuronal Aβ in mouse models and the mystery of how intraneuronal and extracellular pools are related. The cutting edge of this field has advanced past merely trying to detect it inside neurons and toward the issue of oligomeric species (see upcoming SfN news summary). Exactly how its accumulation might damage the neuron has become a burning question. Here, synaptic biology, the role of Aβ in cellular signaling pathways and proteasome degradation, as well as interactions between intraneuronal Aβ and tau, are emerging areas that engage scientists in lively speculation. Selected highlights from the conference follow below in a four-part news series on intraneuronal Aβ. As always, your Alzforum reporters invite kvetches, kudos, and comments.
See also Part 2, Part 3, and Part 4.
Mice Strains Multiply
Research using mouse models to study intraneuronal Aβ goes back a decade. In 1995, Frank LaFerla first reported that a strain of transgenic mice that expressed Aβ inside neurons—but not another strain that expressed it extracellularly—developed pathology and lost neurons (LaFerla et al., 1995). LaFerla followed up with a study on intraneuronal Aβ and p53 activation in mice, and another one showing intraneuronal Aβ in human AD brain. In 2000, Gunnar Gouras at Weill Medical College of Cornell University in New York City and colleagues demonstrated intraneuronal Aβ accumulation in human brain (Gouras et al., 2000), but still, the topic was slow to catch on. Two years later Gouras’s group detected intraneuronal Aβ in young Tg2576 mice (Takahashi et al., 2002), and soon after that the triple transgenic mice produced by LaFerla’s group, then at University of California, Irvine, drew widespread attention (Oddo et al., 2003). At SfN, the knowledge base for this topic broadened when additional investigators introduced their models and showed similar findings.
For example, Robert Vassar, of Northwestern University in Chicago, presented an analysis of his 5XFAD transgenic mice, which coexpress three different AD-causing APP mutations plus two different PS1 mutations driven by the Thy-1 promoter. This genetic combination serves to crank up Aβ42 production so that researchers need not wait 6 months before they can seriously study their mice. In this accelerated model, cerebral Aβ levels rise rapidly beginning at 6 weeks of age, and amyloid deposits begin to appear at 2 months, Vassar reported. At 4 months, learning and memory deficits emerge, and neurons begin to degenerate. On neurodegeneration, Vassar noted that his lab has not yet done definitive stereologic counts, but he has seen synaptophysin levels drop and massive neuronal loss in relevant areas, for example, cortical layer 5 and subiculum. Gliosis accompanies these changes. By 9 months, the mice’s brains are packed with plaques, Vassar reported. Also at this time, the female mice (but not the males) suffer a sudden spike in levels of the proinflammatory cytokine Il-1b and start dying.
More to the point of the topic at hand, however, the Vassar group sees a punctate pattern of intraneuronal Aβ starting at 6 weeks and accumulating with age. (Researchers generally interpret punctate staining as indicating accumulation.) Thioflavin S staining indicates that a portion of this intraneuronal Aβ is aggregated, Vassar said, and some of it appears to occur in neurites. Neurons containing ample Aβ appear to form plaques, Vassar added, leading him to support the hypothesis that aggregating intraneuronal Aβ might cause neurodegeneration and then form the nidus of beginning plaques. It’s worth noting that preliminary studies with tau antibodies have not so far indicated a link between the aggressive Aβ42 production and tau pathology (see ARF SfN tau series), but this question needs more work.
Another new mouse model that features intraneuronal Aβ is that of Lars Lannfelt’s group at Uppsala University, Sweden. Data presented by Lars Nilsson demonstrate that a line that expresses the Swedish APP mutations, which enhance β-secretase cleavage of APP, in addition also expresses the Arctic mutation in Aβ that Lannfelt’s group had identified earlier (Nilsberth et al., 2001). This mutation yields a form of Aβ that is particularly aggressive in forming soluble protofibrils. In these ArcSweTg mice, too, staining for aggregated intraneuronal Aβ predated extracellular Aβ aggregates. It also appeared qualitatively different from the extracellular Aβ, suggesting that aggregates on either side of the cell membrane have structural differences, Lannfelt noted. Aβ deposition in these ArcSwe mutants occurs so rapidly that the mice have compact senile plaques by 5 to 6 months of age. However, there was little evidence of neuronal loss. This data is published online (Lord et al., 2006).
The Lannfelt mice are similar to a strain made in the laboratory of Lennart Mucke at the University of California, San Francisco. These mice also express the human Arctic Aβ mutation plus the Swedish and Indiana mutations in APP, though technical details differ. A brief communication about these mice appeared last year (Cheng et al., 2004), but the paper did not mention intraneuronal Aβ. (Incidentally, Irene Cheng presented further data on these mice at the SfN conference; see upcoming Alzforum news summary on Aβ oligomers.)
Lannfelt’s and Vassar’s mice featuring intraneuronal Aβ follow a model published last year by Laurent Pradier and Thomas Bayer (Casas et al., 2004), but these scientists did not present further data in Washington, D.C.
Do Oligomers Start Forming Inside Neurons?
The LaFerla lab, from the University of California, Irvine, did present a range of follow-up studies of their mice. One of them focuses on intraneuronal oligomerization of Aβ; it appeared online this month (Oddo et al., 2005). The scientists collaborated with William Klein, also at Northwestern, and Charles Glabe, also at UC Irvine, to characterize the time course of Aβ oligomerization in the triple transgenic mice with Klein’s M71/3 (anti-ADDL) antibody, which recognizes 12-24mers of Aβ, and Glabe’s A11, which detects larger species. In brief, the researchers first detect significant amounts of soluble, non-oligomeric Aβ at 4 months of age and Aβ oligomers by 6 months of age, both intraneuronally, in the CA/subiculum region of hippocampus. The researchers suggest that the oligomers begin forming inside neurons between the ages of 2 and 6 months and then accumulate progressively. By 1 year, the presence of Aβ oligomers in this brain region had changed from being intraneuronal to being primarily extracellular and occurring near plaques.
The intracellular oligomers were near the cell body and also in neurites near synaptic sites, Salvatore Oddo reported in Washington. Further experiments indicate that they co-localize with tau in the somatodendritic compartment of 6-month-old triple transgenic mice, but not with hyperphosphorylated forms of tau in old mice. While it is tempting to speculate about an interaction between Aβ oligomers and tau at synapses, this co-localization does not prove it by any means, the scientists caution in their paper. Using a broader panel of Aβ-antibodies, Oddo et al. found that their staining patterns overlapped only partly, suggesting that Aβ occurs in different conformations inside neurons. Together with other data, this supports the notion held by some scientists that separate aggregation pathways for Aβ might exist side-by-side in vivo. This idea further holds that a portion of the Aβ oligomers may be physically stable, rather than fleeting fibrillization intermediates on the way to plaques. This would make them an attractive new target for future therapies.
Finally in this study, the researchers extended previous experiments using anti-Aβ antibodies, which had reduced both Aβ and early tau pathology (Oddo et al., 2004). Trying to target oligomeric Aβ this time, they injected the A11 antibody into the hippocampus of 12-month-old triple transgenic mice and found that it, too, reduced not only intraneuronal Aβ but also tau pathology. This suggests that intraneuronal Aβ oligomers represent a link between Aβ generation and tau, bolstering their status as an attractive target, the scientists write.—Gabrielle Strobel.
See also Part 2, Part 3, and Part 4 of this series.
No Available Comments
This is the second installment of a four-part news series about intraneuronal Aβ from the 35th Annual Conference of the Society for Neuroscience, held November 12 to 16 in Washington, D.C. See also Introduction and Part 1, Introduction and Part 3, and Introduction and Part 4.
Synaptic Activity: It’s Important, But How?
What exactly is Aβ doing inside neurons? Several lines of investigation are converging on some function near synapses. To start this section out with a hint from comparative pathology, consider the presentation by Rebecca Rosen, Lary Walker, and colleagues at Emory University in Atlanta. Using immunohistochemistry, this group compared the localization of intraneuronal Aβ in hippocampus and other brain areas in aging humans, chimpanzees, squirrel monkeys, and rhesus monkeys. All four species deposit Aβ with age, but only humans suffer neurodegeneration as seen in AD. The researchers found that of the four species, only humans had dense Aβ puncta in the dendritic compartments of their hippocampal pyramidal neurons. That’s where synapses reside, and several groups have begun focusing intensely on exactly what Aβ might be doing there.
Helen Hsieh in Roberto Malinow’s lab at Cold Spring Harbor Laboratory, New York, asked if increased levels of Aβ might affect the trafficking of receptor proteins. Previous work from this lab had shown that activity increases Aβ release, and that Aβ then depresses AMPA and NMDA receptor-related neurotransmission in hippocampal synapses (see ARF related news story). At the SfN conference, Hsieh showed two-photon laser scanning images of hippocampal neurons overexpressing APP, which showed that they were less densely packed with synapses. Next, Hsieh tested if Aβ might influence long-term depression (LTD), a mechanism of synaptic plasticity that results from endocytosis-mediated removal of the GluR2 receptor from the synapse. When Hsieh measured transmission in organotypic hippocampal slice cultures from neurons that overexpress APP or β-CTF, she found that transmission was reduced and LTD was decreased compared to wild-type neurons. Overexpression of an APP mutant that produces no Aβ had no effect on LTD. Thus, expression of APP or β-CTF mimicked and occluded LTD. Cotransfecting APP into neurons with a rectifying GluR2 resulted in less rectification, indicating synaptic removal of GluR2. Both sets of electrophysiology measurements suggest that Aβ changes GluR2 trafficking.
To address the fate of the receptors directly, Hsieh measured the level of GluR2 at the synapse and found that it was down in cells overexpressing APP. Furthermore, she reported that both the p38 MAP kinase inhibitor SB203580 and the phosphatase calcineurin, which block metabotropic glutamate receptor LTD, can attenuate the effect of APP. All these results indicate that Aβ increases endocytosis of GluR2 receptors mediated by clathrin-coated pits and in this way depresses synaptic transmission. This conclusion draws further support from Hsieh’s finding that the effects of APP and β-CTF disappear in cells expressing an endocytosis-resistant receptor.
How these experiments—conducted on young hippocampal slices from mice overexpressing APP—relate to AD is unclear at present. Also unclear is how Aβ might influence receptor trafficking. And glutamate receptors2 are not the only candidate victims of intraneuronal Aβ. GluR1 receptors have been reported to be down in Tg2576 mice (see Almeida et al., 2005), and LaFerla’s lab has reported that, in triple transgenic mice, α7 nicotinic acetylcholine receptors disappear from neurons in brain areas where intracellular Aβ oligomers accumulate (Oddo et al., 2005). These latter receptors are thought to interact with Aβ (see prior ARF SfN meeting report) and in Washington, Kelly Dineley from the University of Texas Medical Branch, Galveston, reported that deleting them in Tg2576 mice worsened the mice’s learning and memory deficits.
A fundamental question in this regard is whether Aβ strikes synapses from without or within. Food for thought on this question came from a presentation by John Cirrito in David Holtzman’s lab at Washington University, St. Louis, with colleagues at Lilly Research Laboratories in Indianapolis, and the University of Arizona at Tucson. Cirrito reported on experiments using a microdialysis probe to measure the amount of Aβ in the interstitial fluid (ISF) in the hippocampus (see ARF related news story). Their protocol allows them to take samples every half hour for up to 24 hours in awake, behaving mice. Cirrito reported that when hippocampal neurons in these mice were stimulated with electrical probes, the level of Aβ in the ISF shot up. When he used tetrodotoxin to attenuate normal neuronal activity, ISF Aβ went down. The experiments suggest that neurotransmission and release of Aβ are inextricably linked.
To probe this further, Cirrito treated some mice with tetanus toxin to block the release of neurotransmitter vesicles. This reduced the level of ISF Aβ by 80 percent within 8 hours, he reported. When he treated cultured brain slices with α-latrotoxin (which prompts release of synaptic vesicles) together with postsynaptic inhibitors, extracellular Aβ levels still increased, suggesting synaptic vesicle release alone could lead to increased Aβ release. This latter result, coupled with the fact that Aβ has not been found in synaptic vesicles, led Cirrito to speculate that the link between synaptic activity and ISF Aβ is indirect, perhaps related to vesicle recycling. In this regard, readers may want to revisit data by Brent Kelly, Robert Vassar, and Adriana Ferreira at Northwestern, who reported that Aβ decreases levels of dynamin 1, a protein needed for synaptic vesicle recycling (Kelly et al., 2005).—Gabrielle Strobel and Tom Fagan.
See also Introduction and Part 1, Introduction and Part 3, and Introduction and Part 4 of this series.
No Available Comments
This is the third installment of a four-part news series about intraneuronal Aβ from the 35th Annual Conference of the Society for Neuroscience, held November 12 to 16 in Washington, D.C. See also Introduction and Part 1, Part 2, and Part 4.
Is It the Pits? Aβ and Endosomes
The vesicle-Aβ link found by Cirrito was also the topic of a talk by Claudia Almeida in Gunnar Gouras’s lab. When these scientists reported earlier this year that neurons from Tg2576 transgenic mice had reduced surface expression of the GluR1 variant of glutamate receptor, they also noticed that these losses were accompanied by losses of PSD-95, a protein that anchors GluR1 to the synapse. In Washington, Almeida addressed how Aβ might influence cell surface receptors and proposed that it does so by impinging on the multivesicular body sorting pathway.
Multivesicular bodies, or late endosomes, deliver membrane proteins to lysosomes for degradation, and Aβ42 co-localizes strongly with markers of late endosomes. Early, recycling endosomes shuttle membrane proteins back to the cell membrane, and Aβ42 co-localizes weakly with them. To determine how Aβ42 affects the dynamics of these sorting bodies, Almeida measured trafficking of two markers. The transferrin receptor (TfR) is processed by the recycling of early endosomes, and the epidermal growth factor receptor (EGFR) is shuttled through late endosomes. She exposed neurons from Tg2576 mice to fluorescently labeled transferrin or EGF and then took time-lapse snapshots of their intracellular locale. EGF and Aβ began appearing together after 10 minutes, and did so substantially after an hour, whereas endocytosed TfR did not segregate to Aβ-labeled compartments.
Importantly, Almeida found that in cells expressing hAPP, the recycling EGF appeared to get stuck in late endosomes. In wild-type cells, it was all but gone from endosomes after an hour, whereas in transgenic neurons, both the size and the density of EGF-containing endosomes were significantly larger at this time point. Levels of the phosphorylated, endocytosed EGF receptor stayed up in transgenic cells for much longer than in wild-type. Almeida demonstrated the involvement of Aβ in this process by pretreating cells with the γ-secretase inhibitor DAPT. Under these conditions, phospho-EGFR was inactivated much faster. APP similarly retarded degradation of TrkB receptors through late endosomes. This line of research converges with research by Anne Cataldo and Randy Nixon on the endosomal system and the intraneuronal degradation process of autophagy (see prior SfN news story; Cataldo et al., 2004). Research by Michael Ehlers on how recycling endosomes supply AMPA receptors for synaptic plasticity provides a basic science context on the broader topic (Park et al., 2004).
Recycling of the EGFR requires an intact ubiquitin-proteasome system (UPS); indeed, activity-dependent turnover of many neurotransmitter receptors in dendritic spines is thought to occur through the proteasome (Ehlers, 2003). For this reason, the scientists wondered if Aβ might affect endosomal recycling through actions on the UPS. Almeida reported that neurons from transgenic mice show increased ubiquitination of the EGFR after treatment with EGF. Proteasome function was generally impaired in these neurons, but DAPT treatment restored it to near normal. Furthermore, treating wild-type cells with a proteasome inhibitor slowed the EGFR’s progression through the multivesicular bodies to a crawl, similar to what the scientists saw in APP-transgenic neurons. This suggests that Aβ may influence receptor degradation through actions on the proteasomal system, Almeida said. Other groups also reported hints that Aβ, perhaps its oligomers, impairs proteasome function, potentially slowing down the turnover of receptors and other synapse components.
The results on activity and Aβ available so far highlight a nagging paradox, noted Gouras. Data from Malinow’s, Holtzman’s, and his own lab all indicate that synaptic activity increases Aβ production/release, and increasing Aβ certainly is assumed to drive AD. Yet how does this fit in with the finding that neuronal output gradually declines in the run-up to AD, starting years before the disease is overt? PET imaging and other methods have established the brain’s waning activity. Put most simply, then, the question is: Is thinking good or bad? Epidemiological and other evidence suggests that education and challenging mental activity protect against AD, but it’s also true that one in 10 epileptic patients develop Aβ pathology in areas of excessive activity. Gouras himself is beginning to address the question of how synaptic activity and Aβ might interact to affect pathogenesis through his studies of intraneuronal Aβ. As a first step, his group reported at the SfN conference that when they activated cultured Tg2576 neurons, their intracellular Aβ levels dropped. Perhaps understanding the relationship between intraneuronal and extracellular Aβ could help clear up this mystery, Gouras said.
This relationship is not understood at all. Are the pools on either side of the cell membrane somehow linked, or are they independent of each other? Using the triple transgenic mice, Oddo and colleagues addressed this question with an extension of their previous immunotherapy experiments whereby an anti-Aβ antibody injected into the hippocampus removed the peptide from both the brain parenchyma and from the inside of neurons. In new work presented in Washington, D.C., the scientists showed that the extracellular pool shrinks within 12 hours of the injection, whereas the intracellular pool shrinks after 3 days. Once the antibody has dissipated, the intracellular pool becomes replenished first, and then extracellular Aβ reappears. Moreover, as the triple transgenic mice age, their intracellular Aβ load gradually decreases and extracellular Aβ increases. The authors interpret these data to mean that some sort of dynamic equilibrium connects the two pools of Aβ on either side of the neuronal cell membrane. This work will appear in the American Journal of Pathology next January. In a further suggestion along these lines, a poster presented by Erene Mina from Glabe’s laboratory suggested that autophagy gears up when cultured cells are exposed to extracellular Aβ oligomers from the outside. How this communication works remains an enigma. —Gabrielle Strobel and Tom Fagan.
See also Introduction and Part 1, Part 2, and Part 4 of this series.
No Available Comments
This is the final installment of a four-part news series about the role of intraneuronal Aβ from the 35th Annual Conference of the Society for Neuroscience, held November 12 to 16 in Washington, D.C. See also Introduction and Part 1, Part 2, and Part 3.
Aβ Jams Communication Inside Cell
Besides intracellular trafficking and synaptic activity, there are other areas of cell biology with which intraneuronal Aβ seems to interfere. Henry Querfurth’s group, of St. Elizabeth’s Medical Center in Boston, presented a series of data on how intraneuronal Aβ disrupts neuroprotective signaling and hampers the cell’s stress response. Querfurth reported at the conference, and also in the November 23 Journal of Neuroscience, that intraneuronal Aβ impinges on the phosphatidyl inositol-3 (PI3K)-Akt pathway (Magrane et al., 2005). The serine/threonine protein kinase B known as Akt is established as playing an important role in neuronal survival in normal brain and known to malfunction in conditions ranging from ALS to Huntington disease and schizophrenia. Some labs have even suggested that FAD mutations depress the PI3K-Akt pathway (see, e.g., Ryder et al., 2004). Other bits and pieces of circumstantial evidence for a role of Akt in AD exist in the literature, but they are disparate and do not focus expressly in intraneuronal Aβ.
Using an inducible viral vector system, Jordi Magrane, formerly in Querfurth’s lab, had reported earlier that intraneuronal Aβ42 expression is toxic to cultured neurons (Magrane et al., 2004). This time, Magrane examined the peptide’s possible effect on Akt. Akt itself needs to be phosphorylated to become active, and then it phosphorylates a number of target proteins that go on to perform various neuroprotective functions. Magrane found that intracellular, but not extracellular, Aβ reduced this Akt activation in rat primary cortical neurons. Active Akt puts a damper on the tau kinase GSK3β; in the Aβ-expressing neurons, impaired Akt meant that GSK3β was more active, hinting at a link between Aβ and tau through this channel. The frontal cortex of Tg2576 mice, too, showed reduced phosphorylated Akt compared to wild-type mice at the time that intraneuronal Aβ begins to show up in this model.
On the flip side, overexpressing Akt inside primary neuronal cultures protected the cells from the toxic effects of accumulating intraneuronal Aβ, Querfurth reported. GSK3β is but one downstream target of Akt. In experiments studying another, the scientists found that active Akt induces the chaperone HSP70 and in this way mobilizes the cell’s response to stress. Subsequent siRNA experiments suggested that HSP70, in turn, acts downstream of Akt to protect cells from intraneuronal Aβ.
All together, the scientists suggest that extracellular Aβ, especially synthetic Aβ applied to cultures, may act quite different from intraneuronal Aβ. The inducible adenoviral system this group developed allows them to control the expression of Aβ and then tease out the role of various components and their place in the signal transduction chain, Querfurth said. The researchers believe their system is relevant to what happens in vivo in part because they target Aβ to be expressed in the secretory pathway, mimicking the site where it is made in vivo. It’s still not entirely clear how Aβ throws a wrench into this chain of survival events. Previous work using another cell type had suggested it might interfere with the interaction between Akt and its activator, phosphoinositide-dependent kinase 1 (Suhara et al., 2003), and current research pursues this lead, Querfurth said.
In summary, the scientists propose that in early AD, intraneuronal Aβ might wreak its damage partly by preventing sufficient Akt activation, which would enfeeble the cell’s stress response and steer the cell away from survival signals. Thinking in terms of therapies, this work strengthens the case for neuroprotective strategies trying to induce Akt, as some experimental neuroprotective compounds, such as VEGF, BDNF, and IGF-1, are known to do. For yet a different angle on intraneuronal Aβ presented by this group, see related SfN story on parkin.
Implications for Immunotherapy
Beyond giving a thumbs up to neuroprotective endeavors, the growing body of data on intraneuronal Aβ has implications for ongoing therapeutic approaches in AD. According to Gouras, it raises new questions about how immunotherapies might work. Broadly speaking, scientists studying such therapies consider two hypotheses: Either the antibodies sequester Aβ in the plasma and draw down brain levels indirectly (the peripheral sink hypothesis), or the antibodies cross the blood-brain barrier and bind extracellular Aβ to mark it for ingestion by microglia. Both ideas have experimental support, yet they do not explain why antibodies injected into the brain reduced the levels of intraneuronal Aβ (Billings et al., 2005). Could some therapeutic antibodies even make their way inside neurons? To address this question, Gouras’s group treated APP-transfected neuroblastoma cells and cultured Tg2576 neurons with such antibodies. (Tg2576 cells are a model for this because in these mice’s brains, as in human AD brain, intraneuronal Aβ levels increase with age in multivesicular bodies; see Takahashi et al., 2002.) After a day of incubation with N-terminal anti-Aβ antibodies, the intraneuronal pool of Aβ, and staining in neuronal processes, decreased in both cell types, possibly due to a drop in production, Gouras reported. In these cell cultures, the antibodies not only were internalized into the neurons, but active endocytosis also was necessary for intraneuronal Aβ levels to decrease. Gouras suggests that when the antibody binds to the extracellular, N-terminal domain of Aβ on APP, it might be taken up into the neuron along with APP. Once inside the neuron, the antibody might prevent the effect of Aβ on synaptic and signaling proteins, and in this way ameliorate cognitive deficits independent of extracellular amyloid pathology.
Gouras noted that this new proposed mechanism does not exclude others. Depending on the nature of the immunotherapy (active, passive, choice of antibody, etc.), different mechanisms may be at play. Gouras’s “immunotherapy” of the cultured cells, and also LaFerla’s injections of the triple transgenic mice, used antibody concentrations higher than what may be easily achievable in the brain following peripheral injection. These concentrations were useful to delineate the cellular pathway; testing the in-vivo relevance of this work will be a next step.
In summary, a small but growing chorus of scientists maintains that intraneuronal Aβ, not the extracellular pool that grows at a later stage, plays an early pathogenic role in various AD models, and they speculate that the same process occurs in AD. Extracellular Aβ and amyloid deposition, they purport, later compound the early problems.—Gabrielle Strobel.
No Available Comments
Biomarkers are the buzz of the field these days, and the hum was heard inside the beltway at the 35th Annual Conference of the Society for Neuroscience held in the US capital earlier this month. While the Alzforum has followed developments in AD biomarkers closely (see, e.g., ARF related news story and ARF related Live Discussion), we also keep our noses to the ground for likely portents of other neurodegenerative diseases, particularly Parkinson disease (PD). The Washington meeting featured updates on the search for PD biomarkers, and in these early days, α-synuclein seems to be a frontrunner.
Michael Schlossmacher, Brigham and Women's Hospital, Boston, reported that his lab's efforts to develop antibody-based diagnostics to measure both total and oligomeric-only forms of the protein in the peripheral plasma are bearing fruit (Program No. 13.6). An ELISA (enzyme-linked immunosorbent assay), which uses a sandwich combination of a monoclonal antibody and an affinity purified polyclonal antibody to detect total synuclein, gave good specificity and sensitivity, Schlossmacher reported, while a test designed by his collaborator, Omar el-Agnaf, United Arab Emirates University, Dubai, to specifically detect oligomeric forms does not recognize the monomer (el-Agnaf et al., FASEB J, 2005, in press). In blood from synuclein-null mice "spiked" with α-synuclein, the ELISA can detect over 90 percent of added protein.
This group has begun preliminary tests to evaluate the antibody-based diagnostic and to determine if blood α-synuclein can be validated as a test for PD, multiple system atrophy, dementia with Lewy bodies, or other synucleinopathies. In blinded tests during the development phase, the researchers found that total α-synuclein levels were much higher in two of eight samples received from their collaborator at the NIH, David Miller. These samples turned out to be from the same PD patient, who had a synuclein triplication (see ARF related news story). In a pilot trial, Schlossmacher detected that in samples from 14 living subjects from their Movement Disorder Clinic, there was no statistical difference between total plasma synuclein in six sporadic PD patients and six normal and neurological controls, but levels of synuclein were twofold higher in the two other synucleinopathy patients tested, one with multiple system atrophy and the other suffering from dementia with Lewy bodies. Schlossmacher emphasized that these sample numbers are small and that further testing is required.
However, he also revealed that the parallel assay for oligomeric α-synuclein did detect differences between sporadic PD patients and controls. In all six sporadic PD patients, blood oligomeric α-synuclein was statistically elevated (P Clemens Scherzer, also at Brigham and Women's Hospital, and his group, will analyze the blood synuclein protein levels and peripheral blood mRNA profiles, respectively, in 300 subjects over 2 years.
How α-synuclein, a cytosolic protein, makes its way into the blood is presently unclear. In related work, Valerie Cullen from the same group used the same ELISA test to show that when the gene for α-synuclein was transfected into dopaminergic neural cells in culture, the protein was readily detected in the culture medium. Cullen was unable to detect another cytosolic protein, lactate dehydrogenase (LDH), in the same culture medium. Cullen is currently investigating if lipid binding to α-synuclein, or phosphorylation of it, are relevant to its release into the cell medium.
Continuing the theme of phosphorylation, David Miller, from the National Institute on Aging in Bethesda, Maryland, reported that phosphorylated α-synuclein might serve as a biomarker. Miller and coworkers, including NIA colleagues Mark Cookson and Andrew Singleton, as well as Tamie Chilcote at Elan Pharmaceuticals in South San Francisco, tested both the soluble and insoluble (SDS-solubilized) extracts of brain tissue samples for total (Syn-1 antibody) and serine 129 phosphorylated (Elan’s #11A5 antibody) synuclein. Miller reported that while comparably small amounts of phospho-α-synuclein were found in the soluble fractions of both controls and triplication cases, the amount of phosphorylated protein in the SDS-solubilized fraction was much greater in the extracts obtained from PD tissue than control. The finding suggests that, like tau, synuclein is phosphorylated prior to its aggregation. Miller also reported that he detected phospho-synuclein in blood samples from two patients who have synuclein triplication, and indicated that further studies on blood phospho-synuclein as a possible biomarker are under way.
There may be other molecules that might predict or confirm a diagnosis. Clemens Scherzer reported a proteomics-based approach to find them. Scherzer and colleagues used gene microarrays to get transcriptional profiles of blood samples from PD patients and normal controls. Correcting for blood type and looking for differential expression between normal and PD samples, Scherzer reported that he narrowed the field down from 22,000 potential transcripts to eight that were statistically significant. He reported that using expression values of these eight transcripts allowed him to generate a risk marker score that strongly identified PD patients from a set of 66 subjects that included healthy and disease controls.—Tom Fagan.
No Available Further Reading
In the aftermath of a recent spate of gene discoveries for Parkinson disease, some of the prizes scientists were chasing at the 35th Annual Conference of the Society for Neuroscience, held last month in Washington, D.C., were the identification of parkin substrates and the characterization of the latest gene linked to PD.
The latter, called leucine-rich repeat kinase 2 (LRRK2), is now under heavy scrutiny in several laboratories. For example, Veerle Baekelandt, at KU Leuven, Belgium, reported that LRRK2 mRNA is widely expressed throughout the cerebral cortex of the mouse, as well as in the amygdala and the striatum, an area of the brain that is particularly relevant to Parkinson disease. Weiping Gai and colleagues from Flinders University, Adelaide, Australia, and elsewhere, backed up this finding. These researchers reported immunohistochemical localization of LRRK2 in tissue samples taken from PD, Alzheimer disease (AD), dementia with Lewy bodies, and multiple system atrophy patients. Gai and colleagues found that LRRK2 and α-synuclein co-localized in intraneuronal inclusions, in LB disorders. They also reported that LRRK2 occurs in neurofibrillary tangles in AD samples, though not in plaques. On the other hand, Andrew Grover and colleagues at the Sun Health Research Institute in Sun City, Arizona, reported that LRRK2 antibodies recognize human microglia and that LRRK2-positive microglia are associated with pigmented neurons of the substantia nigra, those same neurons that degenerate in PD.
As for parkin substrates, it appears that we have to hold our breath a bit longer before we find out exactly how this E3 ubiquitin ligase contributes to PD progression. Hints abound, but breakthroughs are not in sight yet. However, Wanli Smith, Peter Pei, and colleagues, working at Christopher Ross’s lab at Johns Hopkins University, Baltimore, Maryland, in collaboration with Ted Dawson’s group, reported a connection between parkin and LRRK2. First, they showed that LRRK2 itself appeared to be predominantly cytoplasmic. When they used antibodies to detect the protein, they found punctate labeling indicative of protein aggregation in a small number of cells (HEK-293 SH-SY5Y, or primary neurons) transfected with the LRRK2 gene. This aggregation increased when parkin or synphilin was coexpressed with LRRK2, Smith reported. He also revealed that in immunoprecipitation studies, LRRK2 and parkin or synphilin (but not DJ-1 or α-synuclein) bound tightly. The researchers also found that expression of parkin led to increased ubiquitination of the aggregates, though not of LRRK2. Mutant LRRK2 caused neurodegeneration in SH-SY5Y cells and in primary neurons. All told, the findings suggest that LRRK2 interacts with parkin and synphilin, suggesting a possible pathogenic pathway, and that mutant LRRK2 is toxic via a gain-of-function mechanism. This work will appear this month in PNAS, Smith et al., 200, in press.
Work from Ted Dawson's and Valina Dawson’s own labs at Johns Hopkins suggest that the aminoacyl-tRNA synthetase cofactor, p38/JTV-1, may be a bona fide parkin substrate involved. Hanseok Ko reported that of all the proposed substrates, only p38 levels were elevated in the brains of both young and old parkin-null mice (see also Ko et al., 2005). Ko reported that p38 is also elevated in brains of patients with familial and idiopathic PD, and that while normal parkin can prevent p38-induced apoptosis, the PD-associated R42P parkin mutant cannot. His poster also revealed that overexpression of p38 in mouse brain leads to loss of dopaminergic neurons in the substantia nigra.
James Palacino, working with Michael Schlossmacher at Brigham and Women’s Hospital, Boston, reported a potential mechanism whereby p38 may mediate parkin toxicity. Palacino found that p38 levels are about threefold higher in brain samples taken from people with autosomal recessive juvenile parkinsonism (AR-JP) than in control samples, though with a single anti-mouse p38 antibody he failed to detect any changes in expression of the protein in brain samples from parkin-null mice. What would be the consequences of elevated p38/JTV1? Because p38 has no ubiquitin E3 ligase activity of its own, Palacino wondered if it cooperates with parkin to regulate the degradation of far-upstream signal element (FUSE) binding proteins, or FBPs. These FUSE binding proteins, of which there are three family members, regulate the expression of c-myc and other genes, and their degradation is known to depend on p38/JTV1.
Palacino looked to see if parkin deficiency affects FBP levels. He reported that both FBP1 and 2, also known as KSRP (KH-type splicing-regulatory protein) are elevated in brain samples taken from both AR-JP patients and parkin-null mice. Next, he checked to see how parkin deficiency might affect transcripts that may be degraded by FBPs. First, he measured steady-state levels of mRNA coding for a slate of about 11 proteins that he previously found were differentially expressed in parkin-deficient mice (see ARF related news story). He reported that none of these levels were altered in parkin-null animals. Next, he turned his attention to the protein tyrosine kinase Src because Src mRNA is known to be degraded in an FBP2-dependent manner. Palacino found that when he transfected parkin into M17 cells, which on their own have little or no endogenous parkin, both FBP1 and FBP2 decreased and that this was accompanied by a significant increase in Src mRNA levels. Also, he reported that Src mRNA levels were over fivefold lower in the brains of parkin-null mice. The data suggest that parkin leads to a reduction in the amount of FBPs, which in turn leads to an increase in the steady-state levels of Src mRNA. Src kinase activity was also much lower in parkin-null animals, he reported.
How these findings relate to PD pathology is unclear, presently. But Palacino pointed out that Src regulates mitochondrial respiration. This process, of course, has been linked time and again to PD through a variety of mechanisms, such as apoptosis, generation of reactive oxygen species, and the toxicity of PD-inducing chemicals such as MPTP. In fact, Src regulates the phosphorylation of a subunit of cytochrome c oxidase, the last redox protein in the mitochondrial respiratory chain, causing an increase in electron transfer. Palacino reported that cytochrome c oxidase activity is low in parkin-deficient M17 cells, but that it doubles when he expresses small amounts of exogenous parkin in the cells. Taken together, the evidence forges a molecular link among parkin, p38/JTV1, and mitochondrial fitness.
Another parkin substrate that has drawn some attention is Pael-R, or parkin-associated endothelin receptor-like receptor. Pael-R was initially identified by Ryosuke Takahashi, RIKEN, in Saitama, Japan, and colleagues as a parkin binding partner. It can induce cell death when overexpression leads to accumulation of misfolded protein. This G-protein-coupled receptor also accumulates in the brains of people with autosomal recessive juvenile onset AR-JP (see ARF related news story). Now, Takahashi and colleagues have developed a Pael-R-based transgenic mouse model that recapitulates one of the major features of PD, namely, the loss of catecholaminergic neurons.
Takahashi and colleagues crossed transgenic mice expressing Pael-R under the regulation of the PDGF promoter with parkin-null animals. Takahashi reported that while parkin-null mice appeared normal, animals that also had one or both copies of the Pael-R transgene exhibited age-related changes similar to those seen in PD patients. At 6 months, for example, the numbers of tyrosine hydroxylase (TH)-positive neurons are similar in all animals, but by 12 months, there is a dose-dependent loss of TH neurons in both the substantia nigra and locus coeruleus of the Pael-R Tg/parkin-/- animals. At 24 months, this loss is more pronounced, and again is greater in the homozygotes than in the hemizygotes. Using immunohistochemistry, the scientists also found that the number of dopaminergic terminals is reduced in the striatum, a major projection site for dopaminergic neurons. Both TH and dopamine transporter (DAT) reactivity tissue is lost in striata from two-year-old transgenics, though the number of striatal neurons does not change as visualized by staining with the neuronal marker NeuN. In addition, Takahashi reported a huge increase (almost fivefold) in GFAP immunoreactivity in the striatum; this indicates rampant gliosis, another pathological feature of PD. In summary, the mice might make a good model for studying dopaminergic degeneration and the role of parkin/Pael-R in that process. It will be interesting to see what behavioral data emerge from studies on these animals.
An entirely different twist on parkin came from the laboratory of Henry Querfurth at St. Elizabeth’s Medical Center in Boston. Charles Moussa’s poster described how tagged wild-type parkin on a lentiviral vector managed to protect neuroblastoma and muscle cells from death induced by overexpression of APP or the β-secretase substrate C-100. At least in this system, Aβ appears to be a parkin substrate. Specifically, parkin ubiquitinated Aβ and reduced its level inside the cells, the researchers suggest.
And last but not least, what about ubiquitination without degradation? Several posters pointed to a curious, parkin-catalyzed ubiquitination that does not affect protein turnover. Darren Moore, from Ted Dawson’s lab, presented evidence that parkin is involved in the multiple mono-ubiquitination of heat-shock protein 70 (Hsp70). Through mass spectroscopy, Moore identified Hsp70 as a parkin binding partner. Mutation in the RING2 domain, or C-terminal end of parkin, abolishes this interaction. Moore reported that the mono-ubiquitination of Hsp70 occurs both in-vitro and in cultured cells. However, inhibiting the proteasome has no effect on the turnover of the ubiquitinated Hsp70, suggesting that the ubiquitination is not necessarily a first step in degradation. Similarly, James Olzmann, from Lian Li’s lab at Emory University in Atlanta, reported that parkin polyubiquitinates DJ-1, which has also been implicated in familial PD. But Olzmann reported that it was specifically the L166P mutant of DJ-1 that gets ubiquitinated, not wild-type protein. In Olzmann’s hands, too, inhibiting the proteasome had no effect on levels of the ubiquitinated L166P DJ-1, indicating that the ubiquitination does not affect degradation of the protein, at least by the proteasome. What role these ubiquitination reactions have, if any, in vivo remains to be determined, but proteasome degradation does not appear to be it.—Tom Fagan.
No Available Comments
No Available Further Reading
For the past decade, Alzheimer researchers have gradually built the argument that small species of the amyloid-β peptide might be harming neurons in ways quite separate from the damage done by fibrillized forms. In short, their slogan is, “It’s Not the Plaques, Stupid!” This research addresses a lingering disconnect between the distribution of plaque pathology and that of cognitive dysfunction in AD, but it has been vexed by the problem that Aβ oligomers were maddeningly elusive. After all, what’s not physically in hand or visible to the eye won’t easily persuade skeptical colleagues. A visit to this year’s 35th Annual Conference of the Society for Neuroscience, held November 12-16 in Washington, D.C., however, made clear that this field is moving rapidly. While there are no breakthroughs to report, the large number of presentations on Aβ oligomers and the frequency with which they come up in conversation with scientists make clear that oligomeric Aβ, also called ADDLs, has become the object of intense investigation.
A major difficulty has been the challenge of obtaining sufficiently pure forms of Aβ for quantitative study. By now, several labs have developed better isolation protocols and tools to characterize various forms of Aβ oligomer. It’s too soon to offer a general comparison between them, and an undisputed lead contender has not yet emerged either for the title of most important pathogenic form, or most attractive therapeutic lead. Yet this area has produced a number of intriguing candidates worth knowing about, and academic labs as well as biotechnology and pharmaceutical companies are exploring experimental vaccines against “their” oligomers. Below are selected summaries of a few such studies. Readers who were at the conference are cordially invited to share their notes with the Alzforum community and write comments on presentations missing from this report.
A group not previously covered in this news section that pursues Aβ oligomer research is Heinz Hillen’s and Ulrich Ebert’s at Abbott GmbH in Ludwigshafen, Germany. Stefan Barghorn presented results of a stable Aβ42 oligomer, which this group calls “globulomer” and believes to be pathogenic. This globulomer joins diffusible oligomers called ADDLs first discovered by Bill Klein (Lambert et al., 1998), and secreted cell-based oligomers studied by Dominic Walsh and Dennis Selkoe (Podlisny et al., 1998) in a growing group of suspects for cognitive deficits in AD. (See also section on Ashe below.) The Abbott poster describes how Hillen and colleagues generated a defined oligomer from synthetic Aβ and fatty acids, and then assessed its possible relevance in vivo. They came up with a 60 kD ball of 12 Aβ molecules in which the middle amino acid stretches (20-42) turn inward to form a hydrophobic ball and the N-termini stick out. Monoclonal (8F5) and polyclonal (5598) antibodies generated against the globulomer bind a structural epitope on it, but not APP, Aβ monomers, or fibrils. The monoclonal antibody stains plaques in sections of human AD and mice overexpressing mutant APP (Tg2576). The scientists report that they have detected the Aβ globulomer epitope in the soluble fraction of the cortex of three AD patients and of Tg2576 mice, but that it is absent from human CSF. Similar to anti-ADDL antibodies, the globulomer antibody binds to processes of cultured hippocampal neurons, but not to glia. This work appeared in print last month (see Barghorn et al., 2005).
Looking for a functional effect of this preparation in brain proved tricky when it turned out that the globulomer does not penetrate tissue well, probably because it is too sticky to diffuse far beyond the site of injection. However, when applied instead to rat brain slices, the globulomer completely blocked long-term potentiation, the scientists reported. Ebert said his group has no evidence that the 60 kD globulomer forms in the human AD brain exactly as the 12mer proposed on the poster. After all, the ingredients of synthetic Aβ and fatty acids sound a bit like a recipe for detergent micelles. Instead, Ebert and colleagues propose that in a person’s aging brain, Aβ might oligomerize only part of the way toward the full globulomer, and perhaps does so in response to intrinsic or environmental risk factors, such as age- or diet-related alterations in membrane composition.
Ebert and Barghorn emphasize two points of their study. First, the structural epitope provides a starting point for immunotherapy. Abbott is pursuing an immunotherapy approach about which the scientists stayed mum. Second, the globulomer is not a way station on a stepwise polymerization of monomer-dimer-oligomer-protofibril-fibril. Instead, they associate on a separate track, of which the globulomer would be a stable product. Some unknown factor may cause a conformational switch that drives Aβ down this pathway, Ebert said.
If pathogenic Aβ oligomers are more than unstable transition states on the way to fibrillization, then it should be possible to isolate them. Indeed, some researchers are beginning to do just that. Karen Hsiao Ashe at the University of Minnesota Medical School, Minneapolis, described the isolation of a pathogenic12mer of Aβ her lab calls Aβ*. At a satellite symposium held by the Alzheimer Research Consortium the day before the SfN conference itself, Ashe described this work as part of her group’s longstanding effort to identify forms of Aβ and tau that cause the cognitive deficit in AD, which Ashe believes to be separate from the deficit that follows structural damage, that is, plaques, tangles, dystrophic neurites, gliosis, and synapse and neuronal loss. To that end, Ashe’s group tried to isolate the molecule that causes the behavioral deficits described in the Tg2576 mice she generated in 1996. Setting a strategy that evokes Robert Koch’s postulates of criteria an agent must fulfill to be considered the cause of a disease, Ashe told the audience she aimed to first identify the causative molecule, then purify it out of brain of impaired mice, and then inoculate young, healthy animals to see if it prompts the memory deficit.
To do this, the researchers first characterized the behavioral deficit of the Tg2576 mice. The mice’s performance declined at 6 months of age to a stable level and then dipped down further at 15 months. The scientists decided to isolate a form of Aβ that tracks this performance and, therefore, searched for one whose level rises at 6 months and then stays unchanged for about 8 months. This ruled out all published species of Aβ, including ADDLs, whose levels rise in this period and do not correlate with cognitive performance, said Ashe. For a while, the project was stuck. But in 2002, Sylvain Lesné joined the lab. He developed a fractionation procedure that pulled, out of a soluble fraction enriched for extracellular components, a highly stable 12mer whose time course over the mice’s lifespan fit the bill. It is a 56 kD complex, confirmed by mass spectrometry master Austin Yang at University of Southern California (see ARF SfN story on tau) to contain a form of Aβ. For the third step of testing its effect in healthy animals, Ashe collaborated with Michela Gallagher at Johns Hopkins University in Baltimore, Maryland, a noted rat behavioral neurobiologist. Full results are not available yet but, in short, injection of Aβ*56 purified from impaired Tg2576 mice into young healthy rats left memory acquisition unchanged but blocked memory retention, Ashe reported. Ongoing steps include a project asking whether the presence of this substance distinguished between people who died with AD from those who died without the disease, and a project focusing on the interaction of Aβ*56 with its receptor.
“This is the first time that an agent that disrupts memory has been purified from the brain of an AD model,” Ashe said. “Because it is the agent that causes the cognitive deficit, this brings us a step closer to develop antibodies that could be specifically used as a diagnostic,” she added. This presentation raised intense interest among the audience. Among the follow-up questions was one about what might make the concentration of one species of Aβ remain stable over 10 months while most other species increase. This work is now in press at Nature. —Gabrielle Strobel.
See also Part 2 of this news update.
No Available Comments
This story continues the Introduction/Part 1 of our SfN conference news update on Aβ oligomers.
The idea that not all oligomers are obligate precursors to fibrils, but instead follow their own, separate pathway has been around for at least a decade, when the first antibodies against such stable species were found, noted Charles Glabe, University of California, Irvine, (see Yang et al., 1995). More recently, it began to gain currency in a broader way. For example, research from other neurodegenerative diseases involving deposition proteins such as α-synuclein and huntingtin suggests that chaperones importantly influence which pathway the monomeric protein will enter (Wacker et al., 2004; Muchowski and Wacker, 2005).
Additional data on Aβ oligomers came from a team at Merck Research Labs in West Point, Pennsylvania. Merck last year licensed Klein’s ADDL technology from the biotechnology company Acumen in South San Francisco, California, and in Washington, D.C., last month, the Merck/Acumen Joint Research Team presented a series of posters and a talk. For example, the scientists developed an assay to compare and quantify how well various antibodies can block the binding of ADDLs to cultured hippocampal neurons. The A11 antibody did not block ADDL binding in this assay, while others, including 6E10 and 4G8 partially blocked it, and the WO-2 antibody, made by Lars Lannfelt’s group while he was still at Karolinska Institute in Huddinge, Sweden, blocked more strongly. Curiously, despite this difference, the immunocytochemical binding of these antibodies to ADDLs looked similar. Cross-binding studies such as these are highlighting that the different groups are each looking at slightly different forms of oligomer. (On this note, the Abbott researchers said that A11 binds their globulomer at 1000-fold lower affinity than do their own antibodies, but Ashe said A11 does recognize Aβ*56.)
Faced with the task of characterizing ADDLs quantitatively, the scientists from Merck/Acumen brought in some analytical prowess. They compared data from standard SDS-PAGE, which had previously indicated the presence of trimers, tetramers, and several other species, with data gathered from solution-based methods such as high-performance size-exclusion chromatography coupled with multi-angled laser light scattering, and with analytical ultracentrifugation and atomic force microscopy. This analysis indicated that ADDLs occur in a much wider range of sizes than previously thought, the team reported.
The Merck researchers also showed data suggesting that ADDL binding to cultured primary hippocampal neurons leads to an increase in phosphorylated tau in those neurons several hours later. This dovetails with data presented by Sally Frautschy and Greg Cole’s group at University of California, Los Angeles. These investigators infused an antibody against the Aβ1-15 epitope into the brains of Tg2576 mice and showed that this experimental passive vaccination reduced not only the levels of Aβ oligomers but also of phosphorylated tau and active GSK3β, one of the kinases known to phosphorylate tau. In subsequent in-vitro and cell-based experiments, Aβ42 oligomer preparations activated GSK3β. The A11 antibody, which recognizes Aβ oligomers but not monomers or fibrils, counteracted that activation. Furthermore, Frautschy showed that CNS infusion of Aβ oligomers stimulated GSK3beta activation and caused cognitive deficits in the Morris water maze, which were blocked by specific GSK3 inhibition. Taken together, these findings strengthen an emerging realization that Aβ oligomers act upstream of tau, and suggest that this might happen in part via GSK3β. They also imply that if a safe and effective vaccine against oligomeric Aβ could be found, it might have an indirect effect on the tau arm of AD pathogenesis, as well. (For summaries of similar experiments by other groups, see ARF SfN tau series and ARF SfN intraneuronal Aβ series.)
The debate around whether plaques/tangles or their smaller predecessors are the primary neurotoxic species in Alzheimer disease has been roiling for years, and it still does. Consider the following two news tidbits, one for each side of the issue. The SfN meeting saw growing evidence suggesting that, at least in transgenic mice, Aβ oligomers are responsible for distinct synaptic and cognitive deficits before mature pathology has deposited. Besides Ashe’s data (see part 1 of this story), conference-goers also noted the work of Irene Cheng in Lennart Mucke’s lab at the University of California, San Francisco. On a poster crowded with visitors, she presented new data on a mouse model of the Arctic mutation that was first discovered by Lannfelt’s group (see ARF SfN story). Residing within the sequence of Aβ itself, this mutation makes the peptide more prone to aggregate. Cheng’s mice expressing this mutation predictably formed neuritic amyloid plaques faster than did transgenics expressing wild-type Aβ (Cheng et al., 2004). At the conference, Cheng showed how she has compared their learning and memory in the Morris water maze to that of mouse strains expressing human wild-type Aβ, which perform poorly in this test. Alongside the behavioral tests, she also compared the concentration, in hippocampal dentate gyrus neurons, of synaptic proteins whose expression depends on the level of activity. These proteins are relatively depleted in the mice expressing human wild-type Aβ. By contrast, mice expressing the Arctic mutation performed normally and had normal levels of the activity-dependent synaptic proteins, even though they had a head full of plaques. There were differences between a low- and high-expressing line, but in a nutshell, the scientists suggest that the onset of cognitive deficits is delayed in a situation where a particularly fibrillogenic form of Aβ drives the peptide into plaque formation so rapidly that relatively fewer of the neurotoxic Aβ oligomers are around to mess with synaptic function.
After barraging our esteemed readers with news on Aβ oligomers, and partly overlapping news on intraneuronal Aβ (see ARF SfN intraneuronal Aβ series), it seems prudent to end on a contrarian’s stance as an antidote to irrational exuberance. Lest oligomerophiles get carried away believing that cognitive dysfunction in Alzheimer disease is all about these small villains, this story will leave them with a final note on plaques. Curious minds may want to stay on the lookout for the publication of an elegant presentation by Edward Stern in Brad Hyman’s group at Massachusetts General Hospital. Stern and colleagues used intraneuronal recordings in live Tg2576 and control mice to address the question of whether plaque deposition distorts the transmission of nerve impulses in cortex. In a prior paper, Stern had recorded from neocortical pyramidal neurons and shown that the synaptic response to stimuli coming from across the corpus callosum was severely impeded when the signal had to travel through plaque-loaded brain tissue, and they had suggested that this might be a functional consequence of dystrophic neurites. Importantly, the cortical neurons were responsive, but plaques distorted their convergent inputs so that the cortical neurons could not integrate them properly (Stern et al., 2004).
In Washington last month, Stern presented a follow-up study in which he took advantage of the whisker barrel system. In this beautiful neuroanatomical array, each whisker on a mouse’s snout has a receptive field that maps first to the thalamus and from there to a distinct column of neurons in the somatosensory cortex. Bending a particular whisker prompts cortical neurons in its column to fire; tweaking neighboring whiskers from within that given receptive field will elicit a weaker yet defined and measurable response. Using this system, Stern and Hyman found that in Tg2576 mice, only the primary whisker prompted a consistent and measurable response in somatosensory cortex. The farther away from the primary whisker Stern stimulated, the more variable and disjointed the response became, in essence narrowing the de facto size of each whisker’s receptive field. In short, the precision and reliability of the cortical response decreased as the distance that convergent inputs had to propagate through plaque-littered areas increased. This possibly occurs as plaques distort the morphology of neurites. One implication is that convergent inputs impinge on their target neurons without the necessary synchrony, and this, in turn, might help explain why association cortex, which depends heavily on integration of inputs from multiple areas, is more affected in AD than is primary motor cortex, for example. Maybe the motto should be: “It’s Both, Stupid!” Final coinage of a slogan will have to await decisive data; meanwhile, pithy suggestions are always welcome.—Gabrielle Strobel.
One poster that stood out at last month’s 35th Annual Conference of the Society for Neuroscience was that of Andre Fischer, Farahnaz Sananbenesi, and colleagues from the labs of Li-Huei Tsai at Harvard Medical School and Bai Lu at NIMH in Bethesda, Maryland. Presenting the latest chapter in the evolving story of what the kinase Cdk5 might do in neurodegeneration and Alzheimer disease, the poster rose above the din of the conference hall by offering a surprising twist that revives an old idea. P25, the small protein that the field has come to view as a pathological activator of the kinase Cdk5, is actually good for learning and memory when it acts briefly and then disappears, the scientists found. Indeed, it might be part of the brain’s compensatory effort at sharpening its cognition in the face of stressors, and turn into a destructive force only when active in runaway fashion. The study appears today in the journal Neuron (Fischer et al., 2005).
In the past 6 years, a series of studies by several labs has placed the serine/threonine kinase Cdk5 amid the intersecting web of hypotheses of what causes AD. Cdk5 is known to be important for brain development and has been implicated in various brain functions in adult life. Late in life, it is suspected of participating in the pathogenesis of AD when its normal, tightly regulated activation by the short-lived protein p35 gets effectively turned into constitutive activation by the long-lived p35 truncation fragment p25. Links to neurodegeneration, and to both major AD pathologies, abound. For example, neurotoxic stimuli induce the protease calpain, which in turn cleaves p35 into p25. The protein tau is among Cdk5’s many substrates, and chronic p25/Cdk5 activation in the forebrain causes neurofibrillary pathology, gliosis, and severe neurodegeneration in mouse models. The connection to APP or the Aβ peptide is less clearly established to date, though some research has placed Aβ upstream of p25. Likewise, high levels of p25 in postmortem AD brain have been described by Tsai’s and one other group but are not widely reproduced as yet.
To sort out what role p25/Cdk5 might play in AD pathogenesis, it would surely help to understand its function. In other words, what is all that phosphorylating good for? The present study addresses this issue by focusing squarely on synaptic plasticity. Prior studies have already implicated Cdk5 in long-term potentiation (LTP) and other aspects of synaptic function. For example, Cdk5 phosphorylates the postsynaptic protein PSD-95 as part of its function in clustering NMDA receptors; PSD-95 also appears to be reduced in the context of accumulating intraneuronal Aβ (see ARF related SfN story; Almeida et al., 2005). Similarly, Cdk5 phosphorylates hippocampal NR2A, an NMDA receptor subunit, as well as other synaptic proteins. To find out how p25/Cdk5 might affect the dynamics of synapses, Fischer et al. used transgenic mice in whose forebrain they could turn p25 production on or off at will by feeding the antibiotic tetracycline or its analog doxycycline. The scientists toggled this switch such that they were able to compare young adult mice that expressed p25 for 6 weeks to others that only made p25 for 2 weeks and then lived another month without it. The researchers assessed cognitive performance, LTP, dendritic spine density, and synapse number.
As expected, Fischer et al. found that prolonged expression of p25 severely impaired LTP in the hippocampus. The mice performed poorly in a fear-conditioning test and the Morris water maze. They also suffered astrogliosis, lost synapses, and their neurons died in droves. The surprise came with the other mice, in whom transient exposure to p25 turned out to be an altogether good thing. They aced the associative and the spatial learning and memory tests, and cultured slices of their hippocampus produced enhanced LTP. They had more dendritic spines on their neuronal dendrites, and more synapses, than control mice. Their neurons stayed alive and well. Intriguingly, their improved performance lasted for a month until after p25 was shut off again, as if its temporary presence had indelibly changed the synapses. The cellular mechanisms for this require further investigation. Early hints and prior work, however, suggest that NR2A phosphorylation and a subsequent facilitation of NMDA receptor signaling may have much to do with the improved LTP. Formation of new spines and synapses, possibly mediated by the Cdk5 substrate PAK-1, could further improve cognitive performance, Fischer and colleagues write.
In summary, the data show that regulated Cdk5 activity is important not only for development but also for learning and memory, writes Frank LaFerla of the University of California, Irvine, in an accompanying news and views article. A critical next step for unraveling the role of Cdk5 in AD pathogenesis will be to analyze how aging mice respond to p25 induction, LaFerla writes. But even now, he concludes, the available body of work on Cdk5 points to this kinase as a central player in memory, plasticity, and the pathologies of AD.
Not all findings in this study fit neatly into a clear-cut picture. The authors point to the paradox that mice with prolonged p25 expression retain high numbers of dendritic spines even as they perform poorly, suffer neurodegeneration, and show electrophysiological deficits. Perhaps this could mean that the additional spines formed in response to p25 induction are not functional. Their increase might reflect the brain’s attempt to compensate for the overall decline in synaptic activity while p25 levels stay high, the authors speculate. Whatever it will turn out to mean, the present data, together with cell culture experiments in this study and prior papers, suggest that p35/Cdk5 normally regulates the formation of dendritic spines, and that p25 stokes this process, the authors note. This would be part of the cellular mechanisms of synaptic plasticity regulated by Cdk5, which can be beneficial with short-term use of p25 but detrimental with chronic exposure.
Finally, the authors leave the reader with some speculation on the broader implications of their data. Could p25 have a physiological role in synaptic plasticity? Other investigators have described evidence for altered neuroplasticity in AD and in reaction to brain injury. Perhaps p25 starts out as a compensatory response to risk factors, but then goes bad? Some evidence exists suggesting that insults such as Aβ, excitotoxicity, and ischemia trigger p25 expression, the authors note. In some people, p25 levels might increase in response to flagging Cdk5 activity with advancing age. A genetic component appeared in the literature recently, when Rosa Rademakers and colleagues at University of Antwerp in Belgium identified an SNP in the Cdk5 gene in some Dutch and Swedish people with familial AD. To test the idea of compensatory p25 production, it will be important to measure p25 levels in patients with MCI but prior to neuronal loss.
P25, then, could be an example for how the very processes of neural plasticity represent an Achilles heel for the thinking, aging brain. The authors point out that this idea goes back to Marek-Marsel Mesulam and Thomas Arendt. These investigators postulated years ago that mechanisms that enhance neural plasticity can become maladaptive when used chronically. In essence, the hypothesis holds that those areas of the brain that show the most plasticity eventually cave in under its burden.—Gabrielle Strobel
No Available Comments
Take a break from Aβ and tau, and consider something completely different: Is it nature or nurture? Societies have mulled this over ever since the concept crept into our collective consciousness. Today, we know there is no simple answer and that human biology and behavior is rarely, if ever, explained by either phenomenon alone. Epigenetics, the study of changes to the genome that, unlike mutations, do not alter the DNA sequence, has exposed just how inextricably linked nature and nurture truly are. It is not surprising then, that the theme served as an epicurean delight at the 35th Annual Conference of the Society for Neuroscience in Washington, D.C., last month, in the form of a symposium of uniformly high quality.
Epigenetics is of interest to a growing number of biologists and clinicians. It offers an explanation for such disparate phenomena as twin discordance (see ARF related news story) and atypical social bonding (see Fries et al., 2005). At the molecular level, it likely plays crucial roles in neurotransmission (Stadler et al., 2005) and learning and memory in mammals (see ARF related news story). It reveals potential mechanisms underlying various neurologic diseases, including schizophrenia (see Dong et al., 2005) and Rett syndrome (see ARF related news story), and it even suggests a means of treatment (see Sharma, 2005). In fact, researchers are currently developing small molecules that perturb methylation and acetylation, two of the most consequential epigenetic changes, in the hope that they may be turned into drugs to treat a variety of diseases, including schizophrenia (see Tremolizzo et al., 2005), and neurodegenerative disorders, such as Huntington disease (see ARF related news story).
To fully understand epigenetics, one must appreciate why it exists. At the Washington symposium, co-chairs Jonathan Pollock and Christine Colvis from the National Institute on Drug Abuse, Bethesda, Maryland, reminded the audience that epigenetics has its roots in adaptation. As organisms become more complex with longer generational times, it becomes harder and harder for them to adapt through natural selection, Pollock suggested. In short, where genetic change cannot keep pace with environmental change, complex organisms have evolved other means to adapt. Specialized tissues such as neurons have arisen to allow organisms to react rapidly to external influences. But that requires that specific subsets of genes be turned on and off in specialized cells. This is where chromatin remodeling enters the picture; epigenetic change, therefore, is a key element of adaptation based on genetic reprogramming.
It is easy to envisage how the apparatus for genetic reprogramming, once it had evolved to facilitate tissue differentiation in multicellular organisms, was co-opted for more immediate benefit, such as the response to environmental stimuli. Frances Champagne, University of Cambridge, England, reported on how something as simple and innocuous as maternal care can begin the process of chromatin remodeling right out of the womb. Work from Champagne’s and other laboratories reveals that rat pups who get licked and groomed the most turn out to do better in tests of learning and memory, such as the Morris water maze. They also have higher levels of both N-methyl-D-aspartate (NMDA) glutamate receptors and synaptophysin, a synaptic marker, in their brain, suggesting molecular adaptation. Well-groomed pups have higher levels of brain glucocorticoid receptor and appear less anxious than pups that get licked less. Notably, long-time Champagne collaborator Michael Meaney and colleagues at Douglas Hospital Research Center, Montreal, Canada, confirmed in 1999 that this pup behavior is epigenetic rather than inherited. If pups born to so-called “low licking/grooming” mothers are instead reared by “high licking/grooming” mothers, they will behave just like their adopted siblings (see Francis et al., 1999).
Champagne and colleagues have since probed epigenetic changes that underlie behavioral differences in rat pups. She found, for example, that the promoter region of the glucocorticoid receptor (GR) has 17 potential methylation sites and that pups born to mothers that lick less have much higher levels of GR promoter methylation, particularly at one specific methylation site, number 16. These methylation differences are not apparent at birth, but gradually emerge after 5 to 6 days of postnatal care. It turns out that these methylation patterns have molecular consequences. They prevent the binding of transcription factors, such as nerve growth factor-inducible protein A (NGFI-A), Champagne revealed.
These epigenetic changes need not be permanent; this is important when one considers epigenetics in terms of disease pathology or drug addiction. Champagne reported that when rats born to low licking/grooming mothers are given the histone deacetylase (HDAC) inhibitor trichostatin A, then by adulthood their methylation pattern changes to that of rats brought up by high licking/grooming mothers.
Acetylation of histones and methylation of DNA are almost as intertwined as the strands of nucleic acids themselves. Champagne showed that hypermethylation of the GR promoter correlates with hypoacetylation of histone H3. Because the HDAC inhibitor promotes acetylation of the H3 histone, demethylation of the GR promoter ensues, followed by increased expression (see Weaver et al., 2004). Indeed, Champagne showed that infusion of trichostatin A into adult rats leads to greater binding of NGFI-A to the GR promoter. She also revealed that directly stimulating methylation by administering methionine, a precursor to the methyl donor S-adenosyl-methionine, can achieve a similar reversal of maternal programming. Some of this work just appeared in the November 23 Journal of Neuroscience (see Weaver et al., 2005).
Epigenetics and Drug Abuse
In a related vein, Arvind Kumar, from the University of Texas Southwestern Medical Center, Dallas, reported how chronic and acute exposure to drugs of abuse can cause epigenetic changes around a variety of promoters. The long-term effects of drug addiction have been explored for some years. It has become apparent that they can lead to changes in gene expression in the brain, particularly in the striatum, where many of the neurons take part in reward pathways that can lead to addiction. But exactly how those changes occur remains mysterious.
Kumar’s work centers on epigenetic changes in rodent striatum in response to either chronic or acute cocaine administration. While acute cocaine induces expression of the transcription factor cFos, chronic exposure to the drug desensitizes cFos levels back to normal and instead leads to the accumulation of a truncated form of FosB (δFosB). Using chromatin immunoprecipitation experiments, or ChIP, Kumar probes the relation between cocaine and covalent modifications—phosphorylation, acetylation, and phosphoacetylation—of histones on target promoter regions in rats.
He reported that acute administration of cocaine leads to a fivefold increase in phosphoacetylation of histone H3 on the cFos promoter, but no change in newly acetylated histone H3. Histone H4 acetylation does increase about threefold, however. Chronic administration of the drug, on the other hand, leads to increased acetylation of histone H3 around the FosB promoter, Kumar reported, but no changes in histone H4. The ChIPs failed to detect any changes in histone acetylation patterns on promoters that do not respond to the drug, such as those for β-tubulin or tyrosine hydroxylase, suggesting that the phosphorylation and acetylation changes are specific to cocaine-induced promoters.
Kumar has also looked at changes occurring around other gene promoters. Expression of the protein kinase Cdk5 (see ARF related SfN story), for example, is unaffected by acute cocaine but is induced by chronic exposure. Would the chromatin pattern mimic those seen for FosB? Kumar found that not only was acetylated histone H3 increased on the Cdk5 promoter, but that δFosB also bound to it under chronic cocaine administration, suggesting that δFosB may play a direct role in cocaine addiction, a possibility that has been hotly debated.
And like Champagne, Kumar and colleagues have modified the addiction pattern using deacetylase inhibitors. Kumar reported that both trichostatin A and sodium butyrate, a competitive inhibitor of HDACs, enhanced histone H3 phosphoacetylation in response to the drug when administered to rats, and that the animals scored much higher on a test that measures the reward component of their response to cocaine. Overexpression of histone deacetylase, on the other hand, dramatically decreased scores in this behavioral test, confirming the importance of histone acetylation in cocaine addiction. Most of this work was just published in the October Neuron (see Kumar et al., 2005). Kumar and colleagues are now using “ChIP on chip” assays, probing chromatin immunoprecipitation samples with DNA promoter chips, or microarrays, in a genome-wide search for promoters that are hyper- and hypoacetylated after chronic cocaine exposure. So far, nearly 85 promoters have been isolated that have increased acetylated H3 and H4 histones following chronic cocaine administration, and 48 promoters where acetylation of H3 and H4 decreased, he reported.
Richard Goodman, Oregon Health Sciences University, Portland, reported his ChIP experiments to find genes that bind to CREB, or cyclic AMP response element (CRE) binding protein. Though it has been known for many years that CREB binds to CRE, one of the quintessential promoter elements, there are still aspects of CRE/CREB biology that are poorly understood. As Goodman pointed out, in PC 12 cells there is a CRE sequence in the promoter for the somatostatin gene, yet neither CREB nor CREB binding protein (CBP) bind to it. This goes to show that CRE binding is not constitutive, Goodman suggested. Instead, it may depend on how the DNA is programmed in individual cells.
Goodman and colleagues use a sophisticated screening method called SACO, or serial amplification of chromosome occupancy, designed to be unbiased, to identify regions of DNA that bind CREB. SACO combines a ChIP experiment with serial analysis of gene expression, or SAGE (see ARF related news story), to identify DNA in chromatin segments pulled down by CREB antibodies. Using this approach, the researchers have identified thousands of potential genes regulated by CREB (see Impey et al., 2004).
In his SfN presentation, Goodman focused on one of these regions, which happens to harbor a non-coding sequence. In fact, about 20 percent of the sequences identified by the SACO experiment did not code for protein, said Goodman. One of the identified sequences lies upstream of a microRNA (miRNA) sequence called miR132. This microRNA is expressed in neurons and its CRE element does bind to CREB. In fact, expression of miR132 increases dramatically when cells are treated with brain-derived growth factor—the response is even greater than that seen for cFos.
So what does miR132 do? Goodman reported that the microRNA strongly induces neurite outgrowth and that antisense miR132 has the opposite effect. By using a prediction algorithm (MIRANDA, see Enright et al., 2003), he and colleagues identified a putative binding site in the 3’untranslated region (UTR) of the gene encoding the GTPase-activating protein, p250GAP. Goodman reported that miR132 inhibits expression of p250GAP and that that, in turn, promotes neurite outgrowth. This is an example of a CREB-regulated miRNA that regulates growth of neurites by responding to extrinsic trophic cues. The work appeared last month in PNAS (see Vo et al., 2005).
As for epigenetics, miR132 and other miRNAs identified in the SACO screen may also bind to other 3’UTRs, hinted Goodman, most notably that for methyl-CpG binding protein 2 (MeCP2), which is mutated in Rett syndrome. Though MeCP2 was initially identified as a transcriptional repressor that binds to methylated DNA, researchers recently showed that it is involved in RNA splicing, as well (see ARF related news story). Because the MeCP2 gene has a very large 3’UTR that possibly binds up to three miRNAs identified in the SACO screen, CREB signaling may mediate RNA splicing and play an important role in the regulation of methylation-linked transcriptional repression.
Epigenetics in Long-Term Memory
Last but not least, Mark Mayford, Scripps Research Institute, La Jolla, California, also addressed CREB signaling in his studies of epigenetic mechanisms in memory formation. Mayford described experiments to address the role of nuclear calcium signaling in the CREB-mediated induction of protein synthesis that is required for long-term memory.
Using the inducible tetracycline promoter system, Mayford and colleagues generated mice that express calmodulin binding protein (CaMBP) only in the nucleus of neurons in forebrain—the system allows expression to be turned off by adding doxycycline to the diet. Mayford checked the system by inducing seizures in adult mice. In control animals, seizures lead to activation of CREB (by phosphorylation on serine 133) in the CA1 neurons of the hippocampus, but this is blocked in the transgenic animals.
In most electrophysiological tests, the CaMBP mice behave just as controls. Excitatory postsynaptic potentials and paired-pulse facilitations are normal, for example. But they do behave differently in learning and memory tests. When CaMBP is on, latency increases in the Morris water maze and the mice show no preference for the quadrant where the hidden platform is located. If the calmodulin inhibitor is off, then the mice do just as well as control animals. Fear conditioning and novel object recognition tests revealed that mice expressing CaMBP have normal short-term memory but impaired long-term memory, Mayford reported.
Epigenetics entered this line of study when Mayford asked what targets Ca2+/calmodulin might activate to facilitate long-term memory. One likely target, Mayford suggested, is CBP, or CREB binding protein. CBP has histone acetyltransferase (HAT) activity and HATs can activate transcription by unraveling the chromatin to expose promoter elements. They have also been implicated many times in learning and memory (see ARF related conference story; ARF related news story. When Mayford and colleagues expressed a dominant-negative CBP, rendered HAT-less by a point mutation, then spatial memory was impaired. Long-term memory declines, while short-term memory is unaltered. Furthermore, trichostatin A, the deacetylase inhibitor used by Champagne and Kumar above, can rescue animals expressing the dominant-negative CBP, showing once again that histone acetylation and chromatin modification play a key role in behavioral response to the environment. For more detailed reading on the symposium, see a monograph prepared in advance by the presenters (Colvis et al., 2005).—Tom Fagan.
No Available Comments
One of the puzzles of Parkinson pathology is why death comes to only a subpopulation of neurons in the midbrain. The dopaminergic neurons in the substantia nigra (SN) and their projections are lost, while neighboring dopaminergic neurons in the ventral tegmental area (VTA) survive. Multiple animal models of PD show this pattern, suggesting that different causative insults converge on a single, as-yet unknown toxic mechanism. A common denominator is mitochondrial dysfunction, and reduced complex I activity and energy depletion are involved in several models. Why these changes culminate in cell death for some neurons but not others has been a mystery (see Alzforum live discussion on this topic), but two recent studies are shedding new light on this issue.
Work from Birgit Liss, Jochen Roeper, and colleagues at Marburg University, Germany, and in Oxford, UK, and Kobe, Japan, lays the blame on selective activation of the ATP-sensitive potassium channel (K-ATP). This voltage-dependent potassium channel effectively acts as a metabolic sensor in that it opens in response to ATP depletion and oxidative stress in cells. Presented at the 35th Annual Conference of the Society for Neuroscience, held last month in Washington, D.C., and published online November 20 in Nature Neuroscience, the study shows that neurotoxic mitochondrial complex I inhibitors activate K-ATP channels in SN dopaminergic neurons, causing the cells to lose their electrical activity. The same toxins have no effect on the channel or the electrophysiology of VTA neurons. Knockout mice lacking the channel are resistant to neuron loss in two different models of PD. This surprising role of the K-ATP channel in neuron death provides a link between energy depletion, oxidative stress, and the selective killing of SN dopaminergic neurons in PD.
Using tissue slices and single cells from adult mice, the researchers established that both SN and VTA dopaminergic neurons express functional K-ATP channels. They have a similar subunit composition, where the Kir6.2 (i.e., the pore-forming) subunit couples with the SUR1 regulatory subunit. Lowering ATP levels activated the channels in both types of neurons, resulting in cell hyperpolarization. But when Liss and colleagues added the mitochondrial complex I inhibitors rotenone or MPP+, the results were strikingly different. In SN neurons, the PD toxins both caused hyperpolarization and a slowing and cessation of spontaneous electrical activity, while the same treatment had no effect on VTA neurons. The effects were due to K-ATP activation, since SN neurons from Kir6.2 knockout mice displayed no sensitivity to the toxins.
The complex I inhibitors were able to activate K-ATP in VTA neurons, provided they were combined with a low concentration of a mitochondrial uncoupler called FCCP. In SN neurons, the opposite occurred: Low concentrations of FCCP prevented channel opening in response to complex I inhibitors. The authors explain this reversal of sensitivity by suggesting that mild uncoupling causes cells to resist K-ATP channel opening in response to complex I inhibition. The data from SN neurons fits with observations that increased mitochondrial uncoupling protects against neurodegeneration in rodent and primate models of PD.
The VTA cells, on the other hand, could possess a mild constitutive uncoupling that protects them from rotenone or MPP+. In support of this idea, Liss et al. show that VTA dopaminergic neurons express higher levels of the uncoupling protein UCP2 than do the SN dopaminergic neurons.
To explore the role of K-ATP channels in cell death in vivo, the researchers turned to Kir6.2 knockout mice. When treated with the PD-inducing regimen of chronic, low-dose MPTP, these mice were completely protected from SN dopaminergic neuron loss. Likewise, crossing the Kir6.2 knockouts with mutant weaver mice, which spontaneously develop a PD-like pathology due to mutations in the Girk2 channel gene, resulted in partial amelioration of SN dopaminergic neuron loss.
Summing up their results, the authors write, “In essence, the convergence of mitochondrial complex I dysfunction and oxidative stress (both of which are present in Parkinson disease) on the activity of K-ATP channels provides a previously unknown candidate mechanism for differential vulnerability of DA neurons that would couple metabolic stress with electrophysiological failure and selective death of SN DA neurons.”
The downstream events that link channel opening to cell death need to be worked out. Meanwhile, the results imply that chronic opening of the K-ATP channel as a result of long-term stress can lead to neurodegenerative disease. By contrast, short-term activation of the channel protects the brain against energy depletion. This channel is known to regulate the energy balance of the organism as a whole via its roles in insulin release and glucose homeostasis. Clinically, the K-ATP channel is an important target in type II diabetes, where incomplete closing impairs insulin secretion. The sulfonylurea channel inhibitors glibenclamide and tolbutamide are widely used to stimulate insulin secretion, and could provide an existing strategy for neuroprotection, the authors note.
The results of this study dovetail with a separate study in another PD mouse model, presented at the conference by Antonio Pisani at Tor Vergata University in Rome, Italy. In an ongoing collaboration, Pisani’s group uses electrophysiology to find out what goes wrong in dopaminergic neurons of DJ-1 knockout mice made in the lab of Jie Shen at Brigham and Women’s Hospital, Boston (see also Goldberg et al., 2005). DJ-1 loss-of-function mutations cause early onset parkinsonism.
In the latest study, Pisani and colleagues challenged the mice with rotenone and deprived them of oxygen and glucose to mimic ischemia. Dopaminergic neurons in substantia nigra of mice lacking DJ-1 proved to be much more sensitive to these stressors than the same kinds of neurons from wild-type mice. K-ATP channels are known to play a major role in the observed electrophysiological response to rotenone. They are also the preferred candidate for mediating the response to oxygen and glucose deprivation, as they are activated when the ATP content falls in conditions of energetic stress. However, when Pisani et al. looked for the mechanism underlying the heightened DA neuron sensitivity, they found that dopaminergic neurons from DJ-1 knockout mice were much more sensitive to the block of the Na/K pump than were other types of neurons in the striatum. In contrast to the K-ATP channel, the Na/K pump is not voltage-dependent. It follows an electrochemical gradient in order to balance the amount of sodium and potassium across the cell membrane, importing two potassium ions for every three sodium ions it extrudes. Regulating homeostasis to avoid sodium overload inside the cell, this pump is not specific to dopaminergic neurons at all. It depends on ATP and collapses when levels of this cellular fuel drop.
Taken together, the studies suggest that a drop in ATP content might have two consequences. It could block the activity of the Na/K pump, impairing ion homeostasis, and activate ATP-dependent K channels. It is something about the regional specificity of this pump that could mediate the SN dopamine neurons’ unique sensitivity to a loss of cellular energy stores, Pisani suggested. The full set of this data is submitted for publication.—Pat McCaffrey and Gabrielle Strobel
No Available Further Reading
Erythropoietin (EPO), produced by the kidney in response to low serum oxygen, is a major stimulant for red blood cell production. Though this glycoprotein has only been medically approved for the treatment of anemia, there are indications that it can do more than just beef up erythrocyte numbers. Studies suggest that the hematopoietic factor can also protect neurons (see Siren et al., 2001), and that it may even improve cognition, properties that are of particular interest to those studying Alzheimer or other neurodegenerative diseases. These findings have spurred clinical trials to evaluate the benefit of EPO in several neurodegenerative paradigms, including stroke and schizophrenia. On November 16, at the 35th Annual Meeting of the Society for Neuroscience in Washington, DC, scientists gathered to address the role of EPO in the nervous system and to discuss its potential therapeutic benefit to both the peripheral and central nervous systems (CNS).
The first red-blooded evidence that EPO may be important in the CNS came in 1995, almost 20 years after the growth factor was purified. Max Gassman and colleagues at the University of Zurich-Irchel, Switzerland, detected EPO, and its receptor, in mouse brain (see Digicaylioglu et al., 1995). This revelation raised many questions, most notably about what a hematopoietic factor is doing in the CNS, given that red blood cells are not produced there. Ten years later, some of those questions have been answered, but pieces to the puzzle are continually being fitted into place.
It is still not fully clear how EPO expression is regulated in the nervous system, for example. At the Washington mini-symposium, Juan Chavez, from the Burke/Cornell Medical Research Institute, White Plains, New York, brought the issue up-to-date. He revealed that in the CNS, control of EPO expression is similar to that in the kidney, which produces the EPO that stimulates erythropoiesis. In the renal gland, expression of the protein is markedly stimulated by another protein called hypoxia inducible factor (Hif), which only survives when serum oxygen is low. When oxygen levels are normal, Hif is rapidly degraded by the kidney’s ubiquitin proteasome system. Chavez and colleagues found that a similar signaling process occurs in the brain, primarily in astrocytes where Hif levels increase under conditions of oxygen/glucose deprivation. However, he also reported that there is a subtle difference between renal and CNS regulation. In the brain, EPO appears to be controlled by just one member of the Hif family, Hif2. This became apparent after Chavez made conditional knockout mice that lose the Hif1 gene. These animals can still make CNS EPO, suggesting that Hif2, found only in endothelial cells and astrocytes, is the major hypoxia inducible factor in the brain (the role of the third Hif member, Hif3, is unclear). In support of this, Chavez reported that Hif2 (but not Hif1) binds to the astrocyte EPO promoter.
These findings are important to consider in the context of neuronal protection. Neurons die within minutes if they are deprived of oxygen, whereas astrocytes can survive for up to two hours in those conditions. But in co-cultures, neurons survive much longer without oxygen, reported Chavez. So how, exactly, does EPO protect neurons and can the mechanism be exploited for therapeutic purposes?
Murat Digicaylioglu and Stuart Lipton, at the Burnham Institute, La Jolla, California, answered the first part of that question several years ago when they showed that pretreatment of neurons with EPO protects them against ischemia-induced apoptosis, or programmed cell death (see ARF related news story). Last year they showed that the need for pretreatment could be dispensed with if EPO was added to cerebrocortical neurons together with insulin-like growth factor 1 (IGF-1), which has anti-apoptotic properties of its own. They also reported that this synergy was mediated by activation of Akt (protein kinase B), which has been implicated in the pathology of Alzheimer disease (see Digicaylioglu et al., 2004 and ARF related news story). At the Washington mini-symposium, Digicaylioglu revealed that this combination therapy also works in vivo. He reported that administering the combination intranasally to animals suffering from experimental stroke lowered infarct volumes and improved cognition. Though EPO is currently in clinical trials for ischemic stroke, these findings suggest that a combination therapy might prove an even better choice.
EPO as a therapeutic may not be restricted to ischemic events, either. Hannelore Ehrenreich, from the Max Planck Institute for Experimental Medicine, Gottingen, Germany, reported results of a clinical trial to measure the potential benefit of EPO in patients with schizophrenia. The rationale for this study was based on work in animal models and in studies of human patients. In mouse models of schizophrenia, for example, recombinant human EPO improved cognition, while immunohistochemical analysis of human autopsy samples revealed that, compared to normal subjects, people with schizophrenia have much higher levels of EPO receptor in both the cerebral cortex and the hippocampus. Together, these findings hinted that EPO supplementation may benefit schizophrenic patients, a suggestion that was supported by imaging studies. The latter demonstrated that EPO is taken up more readily in the brains of those with schizophrenia (see Ehrenreich et al., 2004).
Based on these observations, Ehrenreich and colleagues initiated a proof-of-principle trial. They administered EPO as a neuroprotective “add-on” treatment to patients taking neuroleptics. Out of 44 patients that started the 12-week trial, 39 finished. Ehrenreich reported that the major outcome was dramatically improved cognition—though EPO had no effect on psychic pathology, as judged by scores in the Positive And Negative Syndrome Scale (PANSS), a commonly used test to evaluate schizophrenia patients. Further details of the trial can be found at Ehrenreich’s website.
The results of this small trial suggest that EPO can have a potent neuroprotective effect in humans, slowing or preventing the recently documented neurodegenerative component of schizophrenia (see Thompson et al., 2001). This raises the question of whether the glycoprotein may also benefit patients with other neurodegenerative diseases, such as Alzheimer disease (AD). Only a clinical trial can answer this question, but Ehrenreich and colleagues have just published data showing that in mice, administration of EPO for as little as two weeks prevents the slow neurodegeneration that continues for months after an acute trauma (see Siren et al., 2005). These findings, coupled with the clinical trial data, suggest that the hematopoietic factor can have a profound impact on the neurodegenerative process and might be beneficial in AD or other degenerative diseases. In fact, Wendy Campana, from the University of California at San Diego, and colleagues reported in a poster that EPO receptor mRNA is downregulated in AD brain compared to normal controls. The loss was significant and greatest in those with later-stage disease, who had about 30 percent lower expression of the receptor. The authors also reported that EPO levels are lower in AD patients, too, though not to the same extent. Finally, Campana and colleagues reported that transgenic mice producing human mutant amyloid-β precursor protein have 50-60 percent less EpoR mRNA in the frontal cortex by the time they are six months old. All these findings suggest that EPO might be beneficial for AD patients.
In the peripheral nervous system, too, EPO may turn out to give neurons a boost and, again, this may be dependent on the action of neuronal helper cells. Campana showed several years ago that EPO attenuates apoptosis when it is initiated by damage to spinal axons in rats (see Sekiguchi et al., 2003) and that it can protect dorsal root ganglia neurons against injury in vivo (see Campana and Myers, 2003). In a very recent paper, she reported that injured sciatic nerve fibers upregulate production of endogenous EPO, and that one of the targets for this EPO is its cognate receptor on Schwann cells. In fact, Campana and colleagues reported that these cells, which produce the myelin sheath that surrounds axons, proliferate rapidly if they are treated with EPO (see Li et al., 2005). At the Washington symposium, she revealed that EPO receptors are massively upregulated on the surface of Schwann cells within a minute after addition of the growth factor. These findings are relevant to axonal degeneration diseases such as multiple sclerosis (MS), and, in fact, studies have shown that EPO can lead to improvements in animal models of MS (see, for example, Agnello et al., 2002 and Savino et al., 2005).
The importance of the Schwann cell connection is supported by work from the labs of Sanjay Keswani and Ahmet Hoke (who chaired the mini-symposium) at Johns Hopkins University, Baltimore, Maryland. Their work brings us back full circle to the question of EPO regulation. Last year they showed that Schwann cells produce EPO in response to nitric oxide (NO) (see Keswani et al., 2004). At the symposium, Keswani reported that NO exerts its effects by increasing levels of Hif1 in Schwann cells. This finding appears to mark a major difference between regulation of EPO in the PNS and CNS given Chavez’s findings that Hif2 mediates production of the glycoprotein in CNS astrocytes (see above). But no matter which of the Hif family is involved, it could turn out that EPO acts on the same downstream targets in both central and peripheral nervous systems. Keswani reported that EPO protects peripheral axons in a phosphatidyl-inositol-3-kinase (PI3K)-dependent manner. This is curious, given that Digicaylioglu and colleagues found that Akt, which mediates PI3K signaling, plays a role in EPO/IGF-1-mediated protection of cortical neurons (see above). Even more curious for AD researchers, perhaps, is Keswani’s finding that phosphorylation of glycogen synthase kinase 3β (GSK3β) is essential for EPO’s effects. GSK3β, of course, is well-known as a tau kinase that is silenced by Akt (see ARF related SfN story). The findings give a tantalizing hint that EPO might protect against tau phosphorylation, a major pathological hallmark of AD.—Tom Fagan.
No Available Further Reading
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.