At this year’s meeting, held virtually from March 3-5, FTD researchers reported their latest findings, safely distanced from computers around the world. Their field continues to inch forward despite facing daunting challenges. International cohort studies have charted the tortuous course of this spectrum of diseases, arming researchers with increasingly validated neuropsychological tests and fluid biomarkers that they are starting to put to use in clinical trials.
Merged Consortia Forge Path to Trials in Frontotemporal Dementia
The term frontotemporal lobar degeneration encompasses a broad spectrum of diseases that, to the researcher’s eye, seem marked by inexplicable, and maddening, variability at every turn. From the genetic mutations that cause it, to the neuropathology that damages the brain, to its clinical expressions, FTLD is sinister—and utterly bewildering—in its diversity. And yet, thanks to international cohort studies launched in recent years, researchers are finally gaining a handle on this syndrome. Starting in 2012 with the Genetic FTD Initiative (GENFI) in Europe and Eastern Canada, and followed in 2015 by the Advancing Research and Treatment for Frontotemporal Lobar Degeneration and the Longitudinal Evaluation of Familial FTD Subjects (ARTFL/LEFFTDS), scientists have collaborated on an unprecedented scale. They are charting the clinical progression of the disease and pegging imaging and fluid markers that track its onset and progression. They have tailored cognitive and functional tests to encompass the many symptoms of FTD, and devised physiological measures that link complex social and emotional deficits to faltering circuitry in the brain.
In addition to dozens of papers published in recent months, researchers shared the fruits of their efforts at the International Conference on FTD (ICFTD), held virtually March 3–5. This news series summarizes their progress, starting with the not-so-simple feat of charting the natural course of the disease. Parts 2 to 5 of this series will cover advances in fluid and neuroimaging biomarkers, along with cognitive, functional, and physiological measures that are primed for use in clinical trials. A newly formed international uber-consortium readies participants to enter trials and collaborates with industry to design studies. Clinical trials for carriers of mutations in progranulin are furthest along, with trials aimed at FTD caused by mutations in other genes close behind.
First, a refresher on FTLD or, in clinical parlance, frontotemporal dementia (FTD). In this spectrum of diseases, neurodegeneration afflicts the frontal and/or the temporal lobes of a person’s brain. The specific frontotemporal regions that are affected vary among the syndromes, indeed even among people with the same syndrome. On one end of the spectrum lies behavioral variant FTD. People with bvFTD develop social and emotional problems. They become apathetic, impulsive, loose empathy. Other people develop language disorders, grouped under the umbrella of primary progressive aphasia. They no longer can call up words, or they speak haltingly. Still others develop semantic dementia, where they no longer understand the meaning of words or situations. Movement disorders also lie along the spectrum, including corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and amyotrophic lateral sclerosis (ALS), a motor neuron disease. The lines between these syndromes can be maddeningly blurry, and some people, such as those with bvFTD-ALS, suffer from a combination of them.
With an estimated prevalence of 11 in 100,000 people, FTD is far less common than AD or vascular dementia (Coyle-Gilchrist et al., 2016). Autosomal-dominant mutations are responsible for 25–30 percent of FTD cases. For bvFTD, roughly 40 percent of people harbor a pathogenic mutation, while the majority of PPA, CBD, and PSP cases are sporadic. Therefore, bvFTD patients make up the lion’s share of participants in familial studies of FTD. Of the familial cases, 95 percent carry a pathogenic variant in one of three genes: progranulin (GRN), tau (MAPT), or C9ORF72, while the remaining few carry a pathogenic variant in a handful of other genes. While MAPT mutation carriers accumulate neurofibrillary tangles of tau, carriers of GRN or C9ORF72 mutations are predominantly burdened by deposits of TDP-43. bvFTD can arise from variants in any of the three genes, while MAPT mutations can also cause PSP, GRN mutations can trigger PPA, and hexanucleotide expansions in C9ORF72 can lead to ALS.
There. Sound complicated? Rest assured, it’s not just you.
Amid this seemingly intractable complexity, Jonathan Rohrer of University College London initiated the Genetic FTD Initiative (GENFI). By 2012, Rohrer had been caring for many families with the disease, watching as adult children of his patients approached their own onset. Year after year, family members asked, “Will there be a treatment for me when I develop symptoms?” Until recently, the answer had always been no. “Now, I can actually point to clinical trials on the horizon, or even some that are already enrolling,” Rohrer said.
The shift became possible when Rohrer and other researchers realized that multicenter, international collaborations were the only way to properly investigate FTD—and because patients and their families joined. Today, GENFI includes centers in the U.K., Netherlands, Belgium, France, Spain, Portugal, Italy, Germany, Sweden, Finland, and Canada. It involves more than 100 investigators and 1,000 participants.
On the heels of GENFI, researchers in North America started two similar studies of their own. Called, awkwardly, Advancing Research and Treatment for Frontotemporal Lobar Degeneration and the Longitudinal Evaluation of Familial FTD Subjects, ARTFL created a network of 19 centers to study both sporadic and familial forms of FTLD, while LEFFTDS tapped eight of those centers to more deeply phenotype people with familial forms (Nov 2014 conference news). In 2019, the two merged into one cooperative study, called ALLFTD. Bradley Boeve at the Mayo Clinic in Rochester, Minnesota, and Adam Boxer and Howard Rosen at the University of California, San Francisco, co-direct ALLFTD. At ICFTD , Boeve reported that ALLFTD currently includes more than 1,500 participants who have been evaluated at a total of 2,293 visits.
After GENFI and ALLFTD, other multicenter, longitudinal cohort studies sprang up, such as the Dominantly Inherited Non-Alzheimer’s Disease (DINAD) study in Australia, the New Zealand FTD Genetic Study (FTDGeNZ), and the Research Dementia Latin America (ReDLat) in South America. Recently, these national or regional studies have joined forces in the Frontotemporal Dementia Prevention Initiative (FPI), an effort that will create a global registry of potential clinical trial participants, and collaborate with industry to plan and execute effective clinical trials (see Part 4 of this series for more on FPI).
Nine years and more than 30 publications later, what has GENFI achieved? Take a recent paper that charted the natural history of bvFTD. In the most comprehensive of such studies to date, GENFI researchers led by Barbara Borroni of the University of Brescia, Italy, tracked the progression of behavioral and neuropsychiatric symptoms among 232 people with different genetic forms of the disease (Benussi et al., 2021).
Between 2012 and 2019, 101 of the participants had had at least one follow-up visit, and 35 came for at least two. A handful of participants showed up for seven visits, making for a total of 400 evaluations across the cohort. Because people’s apathy, disinhibition, and lack of insight worsen as their disease progresses, both clinicians and caregivers face an uphill battle getting participants to return to the clinic once they are symptomatic, said co-author John Van Swieten of Erasmus University in Rotterdam. In light of those challenges and the scarcity of people with the disease, those 400 evaluations are hard-won.
Charting Devastation. Rows show how prevalent nine different symptoms became over time among people with bvFTD, tracked separately for 115 carriers of C9ORF72 hexanucleotide expansions, 78 GRN mutation carriers, and 39 MAPT mutation carriers. [Courtesy of Benussi et al., JAMA Network Open, 2021.]
At each visit, study staff assessed participants’ core behavioral and neuropsychiatric symptoms of bvFTD. All core behavioral symptoms—apathy, disinhibition, loss of empathy, compulsive behavior, and hyperorality—emerged across mutation carriers, with apathy being the most common.
Patients with MAPT variants had the most—and the most severe—behavioral disturbances, especially disinhibition and compulsion. Some began to have neuropsychiatric episodes, with anxiety and depression most common among MAPT and GRN carriers, and hallucinations among C9ORF72 carriers. How severe these were, and how that changed over a person’s progression, also differed between the genetic forms. For example, while participants with C9ORF72 expansions became steadily less depressed in their late stages, depression grew more profound in GRN carriers. This suggests that individuals go down different symptom trajectories in the different genetic forms of the disease. This is important to know to estimate a patient’s prognosis, and eventually for clinical trials that gauge the effects of treatments on different mutation groups.
Just as the progression of a person’s disease varies depending on his or her genetic cause, so does the first symptom that crops up. “For one person, it might be an awkward elevator conversation. Someone else might start stealing, while another gets pulled over for speeding,” said Elizabeth Finger of Western University in London, Ontario, at ICFTD. Some people withdraw from conversations with their loved ones, while others voraciously chat up strangers, perhaps even going to far as to caress them on the back, as one woman’s video of her husband revealed.
Finger and colleagues catalogued and categorized these types of initial symptoms among 185 symptomatic GENFI participants, and among more than 1,200 asymptomatic family members, about half of whom carried a pathogenic mutation (Tavares et al., 2020). Caregivers retrospectively reported first symptoms in their symptomatic loved ones. Apathy was the most common initial symptom, followed by disinhibition, memory impairment, and language problems.
Strikingly, even relatives who carried the same mutation often had different first symptoms. Even so, some themes emerged. For example, MAPT mutation carriers tended to lose normal social inhibitions first, while GRN carriers first tended to stumble over language. During the preclinical stage, i.e., in presymptomatic carriers, people with tau mutations were more likely to be moody and wake up a lot at night, while C9ORF72 mutation carriers were slightly more likely to become socially awkward or show odd behaviors, and GRN carriers seemed to have trouble with everyday skills, such as cooking, using appliances, or paying bills.
Notably, none of these early symptoms would warrant a trip to the neurologist, Finger said. This is especially true for people with sporadic forms of the disease who have no indication they are at risk. People without a known family history of FTD typically do not find their way to a neurologist until the disease has progressed well into the symptomatic stage. Instead, the mood and behavior problems drive people to therapists, marriage counselors, or psychiatrists. Like the nurse (with unrecognized FTD) who one day asked a patient to lend her cash, many people with FTD have a history of losing their jobs due to odd behavior or inefficiency at work. “When enough of these events accumulate over the years, that’s when we finally see them in the clinic,” Finger said.
The puzzling range of early symptoms, and their likelihood of being misinterpreted, makes finding objective biomarkers for the disease essential, Finger and other researchers emphasized. In fact, finding and developing robust fluid, neuroimaging, and other types of markers is a central goal of GENFI and ALLFTD. For progress on this front, read Part 2 of this series.—Jessica Shugart
Benussi A, Premi E, Gazzina S, Brattini C, Bonomi E, Alberici A, Jiskoot L, van Swieten JC, Sanchez-Valle R, Moreno F, Laforce R, Graff C, Synofzik M, Galimberti D, Masellis M, Tartaglia C, Rowe JB, Finger E, Vandenberghe R, de Mendonça A, Tagliavini F, Santana I, Ducharme S, Butler CR, Gerhard A, Levin J, Danek A, Otto M, Frisoni G, Ghidoni R, Sorbi S, Le Ber I, Pasquier F, Peakman G, Todd E, Bocchetta M, Rohrer JD, Borroni B, Genetic FTD Initiative (GENFI).
Progression of Behavioral Disturbances and Neuropsychiatric Symptoms in Patients With Genetic Frontotemporal Dementia.
JAMA Netw Open. 2021 Jan 4;4(1):e2030194.
PubMed.
Tavares TP, Mitchell DG, Coleman KK, Coleman BL, Shoesmith CL, Butler CR, Santana I, Danek A, Gerhard A, de Mendonca A, Borroni B, Tartaglia MC, Graff C, Galimberti D, Tagliavini F, Moreno F, Frisoni G, Rowe JB, Levin J, Van Swieten JC, Otto M, Synofzik M, Sanchez-Valle R, Vandenberghe R, Laforce RJ, Ghidoni R, Sorbi S, Ducharme S, Masellis M, Rohrer J, Finger E.
Early symptoms in symptomatic and preclinical genetic frontotemporal lobar degeneration.
J Neurol Neurosurg Psychiatry. 2020 Sep;91(9):975-984. Epub 2020 Aug 7
PubMed.
FTD Fluid Markers for Degeneration: Check. For Pathology: Not Yet.
As the frontotemporal lobar degeneration field approaches an era of prevention and clinical trials, it needs robust biomarkers that herald the onset of symptoms and track the progression of a person’s underlying disease. Those tools are essential to developing effective treatments. Alas, the complexity of this group of rare diseases confounds the search, and international cohort studies such as Europe’s GENFI and North America’s ALLFTD are grappling with the challenge (see Part 1 of this series). Besides sitting for cognitive tests and questionnaires at every visit, participants with FTD and their asymptomatic family members are undergoing repeated lumbar punctures, blood draws, and brain scans for these longitudinal studies. Nearly a decade in, their efforts are starting to pay off in the form of antecedent markers that change preclinically and some that might hold promise for trials. Alas, with regard to disease-specific molecular markers, à la Aβ and phospho-tau for Alzheimer’s: No dice.
In recent papers, and at the International Conference for FTD, held virtually March 3–5, researchers shared some success stories and set their sights on still-elusive fluid markers—those that reveal the neuropathology lurking in the brain. They offered glimpses of theragnostic biomarkers—i.e., those that suggest a treatment may have met its target. In carriers of a C9ORF72 hexanucleotide expansion, poly-GP dipeptides plummeted in the CSF in response to a C9-targeted drug. For GRN mutation carriers, CSF progranulin rose in response to treatment, and a marker of lysosomal dysfunction tracked with symptom onset.
The best-validated fluid biomarker for FTD is a rather generic one: neurofilament light. NfL’s rap sheet dates back a quarter century, showing that this axonal protein trends upward in many neurodegenerative diseases (Rosengren et al., 1996; Alzbiomarker). GENFI and ALLFTD have established that NfL levels rise in both CSF and plasma prior to symptoms, and that plasma NfL reliably distinguishes between symptomatic and asymptomatic people (Sep 2016 news; van der Ende et al., 2019; Illán-Gala et al., 2021).
A newer marker of mayhem is neuronal pentraxin-2. NPTX2 is a synaptic protein whose concentration drops in CSF as synapses are lost in the brain. A recent GENFI paper reported NPTX2 inching downward in presymptomatic mutation carriers who subsequently became symptomatic (van der Ende et al., 2020). Glial fibrillary acidic protein—a marker of astrogliosis—also recently emerged as a promising CSF biomarker, at least among GRN mutation carriers. In them, GFAP was higher in symptomatic patients and tracked with brain atrophy and measures of symptom severity (Heller et al., 2020). John van Swieten of Erasmus University in Rotterdam led GENFI’s NfL and NPTX2 research; he believes pentraxin2 changes before NfL does, though larger studies still need to confirm this.
Van Swieten noted that these biomarker discoveries rest squarely on the shoulders of a small number of initially presymptomatic mutation carriers who develop symptoms while being tracked. So far in GENFI, that is the case for only nine participants. As a group, presymptomatic mutation carriers do not have significantly different levels of NfL, NPTX2, or other biomarkers compared to noncarriers, he said. The correlations will become clearer as the studies increase in size and length, and more people become symptomatic.
In the meantime, Bayesian statistics might help. At ICFTD, Adam Staffaroni of the University of California, San Francisco, reported results from a disease progression model. To span the spectrum, it incorporates biomarker measurements from mutation carriers across FTLD, rather than relying on individual longitudinal data. Staffaroni wove data from ALLFTD and GENFI, on 677 mutation carriers and 372 noncarriers, into his model to get a view of plasma NfL along with a handful of neuroimaging and cognitive markers. Plasma NfL appeared elevated years before symptoms emerged. This was most pronounced in GRN carriers, whose NfL concentrations were an order of magnitude above those of noncarriers a decade before clinical disease started, and ramped up dramatically afterward. Plasma NfL far outperformed neuroimaging or cognitive measures in terms of how dynamically it changed prior to symptoms.
This is not lost on drug developers. The biotech company Alector is putting plasma NfL to use as a screening tool in its Phase 2 and 3 trials of AL001, an anti-sortilin antibody meant to keep progranulin around longer in brain cells. In addition to symptomatic GRN mutation carriers, the company is selecting presymptomatic GRN mutation carriers with elevated NfL, on the assumption they are nearing onset and that a drug effect will be measurable in them. Whether NfL can also serve as a gauge of treatment responsiveness in FTD is unclear. That said, NfL did fall in successful treatment trials of spinal muscular atrophy and multiple sclerosis (Olsson et al., 2019; Sejbaek et al., 2019).
How about mutation-specific markers to track target engagement in clinical trials aimed at different forms of familial FTD? In carriers of C9ORF72 hexanucleotide expansions, poly-GP dipeptides that get translated off these repeats are indeed detectable in the CSF (Meeter et al., 2018). What’s more, the dipeptides plummet in response to C9-targeted treatment, according to data presented at ICFTD by Adam Boxer, University of California, San Francisco.
Dipeptide Pilot. In response to infusions (blue arrows) with a C9ORF72 -targeted antisense oligonucleotide therapy called afinersen, CSF poly-GP concentration plummeted, while symptoms remained stable, in a man with ALS. [Courtesy of Research Square preprint platform.]
At ICFTD, Boxer presented preliminary data, currently posted on the internet in preprint form, from Robert Brown’s group at the University of Massachusetts in Worcester (Tran et al., 2021). In a pilot experiment, the researchers treated a C9ORF72 expansion carrier who had atypical ALS with a C9ORF72-targeted anti-sense oligonucleotide. Called afinersen, this ASO is meant to silence expression of expanded C9ORF72 allele (Liu et al., 2021).
In response to intrathecal infusion of this ASO, poly-GP dropped in the man’s CSF by about 80 percent. This marker remained low, while the patient’s symptoms remained stable, over more than a year of eight infusions between August 2019 and October 2020. Curiously, the man’s CSF NfL levels rose, only dipping back down toward baseline levels once infusions were spaced further apart. The scientists suggested that this uptick in the neuronal injury marker might reflect nerve irritation provoked by the infusion.
In GRN mutation carriers, the CSF progranulin concentration serves as a biomarker. It is roughly half that in noncarriers, because these heterozygous mutations douse expression of the gene. Data from Alector’s trials suggests their anti-sortilin antibody restores progranulin levels to normal, and also reduces levels of plasma NfL (Aug 2020 conference news; Haynes et al., 2020).
In addition to progranulin itself, markers of lysososmal dysfunction resulting from progranulin deficiency might serve as disease markers, according to data presented by Isabelle Le Ber of Pitié-Salpêtrière University Hospital in Paris. Progranulin is processed into its active forms—granulin peptides—only within the lysosome. But progranulin does not travel to the lysosome unaccompanied; rather, it links up with prosaposin, another protein that relies on lysosomal cathepsins to unleash its active forms, saposins A, B, C, D.
At ICFTD, Le Ber explained that when progranulin is in short supply—as in GRN mutation carriers—fewer prosaposin proteins make it to the lysosome, leading to a build-up of the lysosphingolipids (lysoSPLs) that they process (Zhou et al., 2017). These excess lipids can be detected in the plasma of people with lysosomal storage disorders (Polo et al., 2019). At ICFTD, Le Ber reported that plasma lysoSPL was significantly elevated among 101 symptomatic GRN carriers compared to 27 presymptomatic carriers or 70 controls. The findings raise the possibility that the lysosomal marker could track with disease progression in GRN carriers.
Perhaps surprisingly, mutation-specific biomarkers have remained elusive for MAPT-FTD. In AD, several different plasma and CSF species of phospho-tau are wildly successful these days; alas, to date, no fluid or imaging tools detect the forms of tau that accumulate in FTLD. On the contrary, p-tau biomarkers that help diagnose AD are used to rule out AD in people with FTD. This is somewhat helpful given that people with FTD are commonly misdiagnosed as having AD (Mar 2020 news). Still, FTD researchers really would like their own tau markers to rule in FTD. At ICFTD, Henrik Zetterberg, University of Gothenburg, Sweden emphasized the need for such markers. So far, he confirmed, only the amyloid-dependent form of tau aggregation that happens in AD is detectable in body fluids.
A biomarker for FTLD-tau would be especially useful in cases of sporadic FTLD, roughly half of which have underlying tau pathology. Without a causal mutation, clinicians currently have no way to tell whether a given person with sporadic FTD has tau or TDP-43 pathology in their brain. Such neuropathological biomarkers are the Holy Grail for sporadic FTD, Boxer said. “Now we can rule out AD with a blood test. If we could identify who had underlying tau pathology and who had TDP-43, then we would be in great shape for clinical trials in sporadic FTD,” he said at ICFTD.
Zetterberg said that it will be challenging to develop biomarkers for TDP-43. In people with FTD, this protein ditches its home in the nucleus and accumulates in the cytoplasm. Recent studies have demonstrated that, similar to other proteopathic proteins such as tau and α-synuclein, TDP-43 can behave like a prion, propagating by way of templated misfolding (Jul 2010 news; Oct 2018 news). Taking advantage of this attribute, a recent study used real-time quaking-induced conversion (rtQuIC)—a method that detects prion-like seeds—to detect misfolded TDP-43 in the CSF of people with ALS or FTD who had TDP-43 pathology (Scialò et al., 2020; Orrú et al., 2011).
Similar tests are able to detect α-synuclein seeds in people with PD, tau in people with Pick’s disease, and, of course, prion proteins in people with prion disease (Oct 2020 news; Saijo et al., 2017; Dec 2016 news).
This type of assay requires a high level of expertise to do properly; still, Zetterberg was optimistic that something like it could pick up TDP-43 pathology. Boxer expressed interest, as well, though he noted that similar assays for tau would need to become more sensitive to be useful. Zetterberg and Boxer agreed that these markers might work to distinguish between people with tau versus TDP-43 pathology, yet might not be quantitative enough to track progression of pathology or response to treatment. —Jessica Shugart
van der Ende EL, Meeter LH, Poos JM, Panman JL, Jiskoot LC, Dopper EG, Papma JM, de Jong FJ, Verberk IM, Teunissen C, Rizopoulos D, Heller C, Convery RS, Moore KM, Bocchetta M, Neason M, Cash DM, Borroni B, Galimberti D, Sanchez-Valle R, Laforce R Jr, Moreno F, Synofzik M, Graff C, Masellis M, Carmela Tartaglia M, Rowe JB, Vandenberghe R, Finger E, Tagliavini F, de Mendonça A, Santana I, Butler C, Ducharme S, Gerhard A, Danek A, Levin J, Otto M, Frisoni GB, Cappa S, Pijnenburg YA, Rohrer JD, van Swieten JC, Genetic Frontotemporal dementia Initiative (GENFI).
Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study.
Lancet Neurol. 2019 Dec;18(12):1103-1111.
PubMed.
Illán-Gala I, Lleo A, Karydas A, Staffaroni AM, Zetterberg H, Sivasankaran R, Grinberg LT, Spina S, Kramer JH, Ramos EM, Coppola G, La Joie R, Rabinovici GD, Perry DC, Gorno-Tempini ML, Seeley WW, Miller BL, Rosen HJ, Blennow K, Boxer AL, Rojas JC.
Plasma Tau and Neurofilament Light in Frontotemporal Lobar Degeneration and Alzheimer Disease.
Neurology. 2021 Feb 2;96(5):e671-e683. Epub 2020 Nov 16
PubMed.
van der Ende EL, Xiao M, Xu D, Poos JM, Panman JL, Jiskoot LC, Meeter LH, Dopper EG, Papma JM, Heller C, Convery R, Moore K, Bocchetta M, Neason M, Peakman G, Cash DM, Teunissen CE, Graff C, Synofzik M, Moreno F, Finger E, Sánchez-Valle R, Vandenberghe R, Laforce R Jr, Masellis M, Tartaglia MC, Rowe JB, Butler CR, Ducharme S, Gerhard A, Danek A, Levin J, Pijnenburg YA, Otto M, Borroni B, Tagliavini F, de Mendonca A, Santana I, Galimberti D, Seelaar H, Rohrer JD, Worley PF, van Swieten JC, Genetic Frontotemporal Dementia Initiative (GENFI).
Neuronal pentraxin 2: a synapse-derived CSF biomarker in genetic frontotemporal dementia.
J Neurol Neurosurg Psychiatry. 2020 Jun;91(6):612-621. Epub 2020 Apr 9
PubMed.
Heller C, Foiani MS, Moore K, Convery R, Bocchetta M, Neason M, Cash DM, Thomas D, Greaves CV, Woollacott IO, Shafei R, Van Swieten JC, Moreno F, Sanchez-Valle R, Borroni B, Laforce R Jr, Masellis M, Tartaglia MC, Graff C, Galimberti D, Rowe JB, Finger E, Synofzik M, Vandenberghe R, de Mendonca A, Tagliavini F, Santana I, Ducharme S, Butler CR, Gerhard A, Levin J, Danek A, Frisoni G, Sorbi S, Otto M, Heslegrave AJ, Zetterberg H, Rohrer JD, GENFI.
Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia.
J Neurol Neurosurg Psychiatry. 2020 Mar;91(3):263-270. Epub 2020 Jan 14
PubMed.
Illán-Gala I, Lleo A, Karydas A, Staffaroni AM, Zetterberg H, Sivasankaran R, Grinberg LT, Spina S, Kramer JH, Ramos EM, Coppola G, La Joie R, Rabinovici GD, Perry DC, Gorno-Tempini ML, Seeley WW, Miller BL, Rosen HJ, Blennow K, Boxer AL, Rojas JC.
Plasma Tau and Neurofilament Light in Frontotemporal Lobar Degeneration and Alzheimer Disease.
Neurology. 2021 Feb 2;96(5):e671-e683. Epub 2020 Nov 16
PubMed.
Imaging Exposes Hugely Heterogeneous Brain Changes Among FTDs
Frontotemporal lobar degeneration, as its name implies, involves shrinkage of the frontal and temporal lobes of the brain. Hidden beneath this moniker is a devilish complexity and variability. In some people with FTD, the right side of the brain contracts; in others, the left. Some have more degeneration in temporal areas, while frontal areas fade in others. In some, deep subcortical structures also shrivel. In response to the rarity and heterogeneity of every aspect of FTLD, large international cohort studies have applied a barrage of neuroimaging techniques to investigate patterns of atrophy, white-matter erosion, and breakdown of neural circuitry in the brains of people with sporadic and familial forms of FTD. While this work made it clear that structural changes start years before symptoms in all FTDs, these changes and their trajectories are far from the same, depending on the offending mutation with which an individual is saddled. Partly for this reason, the initiatives’ brain imaging research has not yet come up with robust, easy-to-use neuroimaging measures. Importantly, the field also lacks molecular tracers that illuminate the identity and distribution of neuropathology in the FTD brain.
At the International Conference on FTD, held virtually March 3–5, and in recent publications, researchers compared their latest insights in grappling with this complexity. They described how the brain changes differently across genetic forms of the disease, and closed with the hope that their findings will guide the use of neuroimaging markers in therapeutic trials, most of which are geared toward individual mutation groups.
One of the largest longitudinal imaging efforts in familial FTD to date was conducted by the Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia (LEFFTDS) studies, now united under the name ALLFTD. The observational study engages 19 centers in the United States and Canada, where participants regularly submit to neuroimaging sessions. Rather than deciding a priori which region of the brain might shrink, the researchers use serial imaging data to detect any region in a person’s brain that has shrunk between one session in the scanner and the next. Using this approach and some Bayesian statistics, Adam Staffaroni of the University of California, San Francisco, and colleagues tracked the rate of brain atrophy across the brain among 100 mutation carriers and 60 relatives who were noncarriers (Staffaroni et al., 2020). In a nutshell, each type of mutation exacted its own style of wreckage on the brain.
Using worsening scores on the CDR-NACC-FTLD (see Part 1 of this series) as a gauge of disease progression, the scientists reported that in 28 MAPT mutation carriers, the temporal lobes on both sides of the brain started gradually shrinking in the presymptomatic years. Alas, as symptoms emerged and worsened, the shrinkage revved up and engulfed the frontal lobes. For 33 GRN mutation carriers, atrophy rates in the frontal and temporal lobes held steady in the presymptomatic and mild symptomatic stages of the disease, then ramped up steeply as disease got severe. C9ORF72 carriers were markedly different. For them, frontotemporal atrophy marched onward at constant clip throughout all stages of disease; it appeared untethered to symptom severity. “Although the destiny for the brain in C9ORF72 is similar to that of other pathogenic variants, the path to this point is different, being slower and more constant over time,” the authors wrote.
Avenues to Atrophy. Rates of atrophy across the brain, ranging from mild (light blue) to severe (yellow), were mapped for carriers of pathogenic mutations in MAPT (top), GRN (middle), and C9ORF72 (bottom), across disease stage. CDR+NACC-FTLD score of zero = presymptomatic, 0.5 = mild/questionable, 1 = symptomatic. [Courtesy of Staffaroni et al., JAMA Network Open, 2020.]
This has implications for prognosis as well as for the use of structural MRI as an outcome measure in clinical trials, the authors wrote. At ICFTD, Howard Rosen of UCSF, who led the study, discussed how unbiased imaging could be applied to track atrophy and predict symptom onset in an individual person, regardless of where degeneration started inside his or her brain. Other researchers asked how many serial measurements would be required, and how far apart they would need to be spaced, to provide useful information about progression, for example, in the context of clinical trials. Rosen said he is investigating these questions in the ALLFTD cohort.
Other neuroimaging studies have focused on one gene at a time. At ICFTD, Suzee Lee, also at UCSF, gave an update on her work with carriers of a pathogenic MAPT variant. For all symptomatic carriers, shrinkage was apparent in the frontotemporal cortex, insula, and striatum, whereas one in five presymptomatic carriers had atrophy in the medial temporal lobe, which started as early as their 30s. Lee also examined the integrity of white-matter tracts in the cohort, reporting erosion of the corpus callosum and uncinate fasciculus in symptomatic carriers, a finding that appeared among few presymptomatic carriers (Chu et al., 2021).
At ICFTD, Lee reported new findings using resting-state functional MRI. rsfMRI measures neural activity and gauges how robustly networks in the brain are functionally connected. Lee saw that the connectivity of several brain networks known to be affected in FTD was already changing in people as young as their teens. For example, in young MAPT mutation carriers, connectivity was reduced in the salience network. Dysfunction in this network is thought to underlie many of the core behavioral and emotional symptoms of FTD, including loss of empathy and awkward social interactions. Because abnormally low connectivity in this network was seen at such a young age, Lee proposed that neuronal circuitry might even develop differently in MAPT carriers. Lee can’t address the question directly because children are not participating in ALLFTD.
The new findings dovetail with Lee’s previous work describing how distinct patterns of neuronal circuitry were going awry in GRN and in C9ORF72 mutation carriers (Lee et al., 2019; Lee et al., 2017). Interestingly, while connections tended to wane in key neural networks in presymptomatic MAPT and C9ORF72 carriers, some of those same networks became hyperconnected in GRN carriers. The finding is yet another example of the bewildering variability of FTD.
Lee and other researchers are also using multimodal MRI; in other words, they are examining changes in gray-matter volume and white-matter tracts in an effort to more accurately predict symptom onset. Some studies have found that crumbling circuitry precedes gray-matter atrophy in familial FTD, predicting symptom onset within four years (Jiskoot et al., 2019; Feis et al., 2020). Alas, John van Swieten of Erasmus University in Rotterdam, who led this work, said that while shrinkage of the brain does happen in FTD’s presymptomatic phase, the profound variability in terms of which regions shrink when, and how this speeds up relative to symptoms, makes MRI less robust, and less practical, for biomarker use than fluid markers such as plasma NfL.
Ultimately, the most sensitive tools to predict symptom onset and track progression will combine fluid, neuroimaging, and cognitive measures, van Swieten believes. A recent cross-sectional GENFI analysis that modeled a cascade of biomarker changes in GRN mutation carriers exemplifies this approach (Panman et al., 2021). The study included 56 presymptomatic and 35 symptomatic GRN variant carriers, including people with PPA and bvFTD. The authors reported that language problems and plasma NfL rose first, followed by loss of integrity in specific white-matter tracts, and then gray-matter atrophy.
Curiously, for people with PPA, this sequence of changes mapped neatly onto functional and cognitive measures of disease progression, including the FTD-CDR-SOB, a modified version of the CDR-SOB that includes behavioral and functional symptoms specific to FTD. In contrast, for people with bvFTD, the biomarker cascade was only loosely tied to clinical disease severity, once again underscoring the obstreperous heterogeneity that is part and parcel of FTD.
This ICFTD was noticeably devoid of PET imaging data, which have taken Alzheimer’s research conferences by storm for many years. The FTD field still lacks molecular tracers that reliably bind to the different forms of tau that accumulate in people across the FTD spectrum, much less to the TDP-43 pathology that accumulates in about half of people with bvFTD or PPA. Some up-and-coming tracers do bind to the 4R-tauopathies in people with PSP or CBD (Jul 2020 news), but for people with sporadic FTD, the molecular identity of the neuropathology lurking in the brain cannot at present be determined during life.
Along with fluid and imaging markers, the FTD field is designing cognitive, functional, and physiological tests that measure clinical symptoms of the disease. Read the next part of the series to hear more about progress on this front.—Jessica Shugart
Panman JL, Venkatraghavan V, van der Ende EL, Steketee RM, Jiskoot LC, Poos JM, Dopper EG, Meeter LH, Donker Kaat L, Rombouts SA, Vernooij MW, Kievit AJ, Premi E, Cosseddu M, Bonomi E, Olives J, Rohrer JD, Sánchez-Valle R, Borroni B, Bron EE, Van Swieten JC, Papma JM, Klein S, GENFI consortium investigators.
Modelling the cascade of biomarker changes in GRN-related frontotemporal dementia.
J Neurol Neurosurg Psychiatry. 2021 May;92(5):494-501. Epub 2021 Jan 15
PubMed.
Further Reading
Papers
Malpetti M, Jones PS, Tsvetanov KA, Rittman T, van Swieten JC, Borroni B, Sanchez-Valle R, Moreno F, Laforce R, Graff C, Synofzik M, Galimberti D, Masellis M, Tartaglia MC, Finger E, Vandenberghe R, de Mendonça A, Tagliavini F, Santana I, Ducharme S, Butler CR, Gerhard A, Levin J, Danek A, Otto M, Frisoni GB, Ghidoni R, Sorbi S, Heller C, Todd EG, Bocchetta M, Cash DM, Convery RS, Peakman G, Moore KM, Rohrer JD, Kievit RA, Rowe JB, Genfi GF.
Apathy in presymptomatic genetic frontotemporal dementia predicts cognitive decline and is driven by structural brain changes.
Alzheimers Dement. 2021 Jun;17(6):969-983. Epub 2020 Dec 14
PubMed.
Olney NT, Ong E, Goh SM, Bajorek L, Dever R, Staffaroni AM, Cobigo Y, Bock M, Chiang K, Ljubenkov P, Kornak J, Heuer HW, Wang P, Rascovsky K, Wolf A, Appleby B, Bove J, Bordelon Y, Brannelly P, Brushaber D, Caso C, Coppola G, Dickerson BC, Dickinson S, Domoto-Reilly K, Faber K, Ferrall J, Fields J, Fishman A, Fong J, Foroud T, Forsberg LK, Gearhart DJ, Ghazanfari B, Ghoshal N, Goldman J, Graff-Radford J, Graff-Radford NR, Grant I, Grossman M, Haley D, Hsiung G, Huey ED, Irwin DJ, Jones DT, Kantarci K, Karydas AM, Kaufer D, Kerwin D, Knopman DS, Kramer JH, Kraft R, Kremers W, Kukull W, Lapid MI, Litvan I, Mackenzie IR, Maldonado M, Manoochehri M, McGinnis SM, McKinley EC, Mendez MF, Miller BL, Onyike C, Pantelyat A, Pearlman R, Petrucelli L, Potter M, Rademakers R, Ramos EM, Rankin KP, Roberson ED, Rogalski E, Sengdy P, Shaw LM, Syrjanen J, Tartaglia MC, Tatton N, Taylor J, Toga A, Trojanowski JQ, Weintraub S, Wong B, Wszolek Z, Boxer AL, Boeve BF, Rosen HJ, ARTFL and LEFFTDS consortia.
Clinical and volumetric changes with increasing functional impairment in familial frontotemporal lobar degeneration.
Alzheimers Dement. 2020 Jan;16(1):49-59. Epub 2020 Jan 6
PubMed.
Tsvetanov KA, Gazzina S, Jones PS, van Swieten J, Borroni B, Sanchez-Valle R, Moreno F, Laforce R Jr, Graff C, Synofzik M, Galimberti D, Masellis M, Tartaglia MC, Finger E, Vandenberghe R, de Mendonça A, Tagliavini F, Santana I, Ducharme S, Butler C, Gerhard A, Danek A, Levin J, Otto M, Frisoni G, Ghidoni R, Sorbi S, Rohrer JD, Rowe JB, Genetic FTD Initiative, GENFI.
Brain functional network integrity sustains cognitive function despite atrophy in presymptomatic genetic frontotemporal dementia.
Alzheimers Dement. 2021 Mar;17(3):500-514. Epub 2020 Nov 20
PubMed.
Moving Target: Can Standardized Tests Track Symptoms of FTD?
For people with FTD and their caregivers, the disease takes a deep personal toll, straining the social and emotional connections at the core of their relationships. Mirroring the diversity of human personality and behavior, the symptoms of FTD, which is actually a group of syndromes, are remarkably varied, spanning social, emotional, executive, behavioral, language, and even motor dysfunction. How can researchers measure these symptoms objectively, let alone encapsulate them within standardized tests? The challenge is real, but a necessary one to tackle, as the field moves into an era of clinical treatment and even prevention trials. Such measures will need to be sensitive enough to find out whether an investigational drug worked in a clinical trial, yet inclusive enough to encompass FTD’s clinical manifestations.
Because FTD is rare and complicated, large international consortia, particularly Europe’s GENFI and North America’s ALLFTD, have been key to devising and validating such tests (see Part 1 of this series). Composite measures that detect an ever-widening swath of deficits are beginning to be put to use in clinical trials, while new measures of social and emotional cognition—from a “faux pas detector” to eye tracking—are opening windows to specific neuronal circuits that malfunction in the disease (see Part 5 of this series). These clinical tests will be used in conjunction with fluid and imaging biomarkers to track the course of the disease (see Part 2 and Part 3).
In a flush of recent papers, and at the International Conference for FTD, held virtually March 3–5, researchers described their attempts to wrestle the breadth of FTD symptoms into standardized tests, as well as their explorations of the neurophysiology of those symptoms.
One low-hanging fruit in the search for clinical tests in FTD has been to pick off tools already in place for Alzheimer’s. In that disease, the clinical dementia rating scale sum of boxes (CDR-SB) assesses impairment by tallying deficits in six cognitive and functional domains. To tailor it to FTLD, researchers long ago added two domains for language and behavior (Knopman et al., 2008). The amended version got the pithy name “CDR plus National Alzheimer’s Coordinating Center (NACC) Behavior and Language Domains”, aka CDR plus NACC FTLD. This composite measure weighs all eight domains equally, whereas the memory-centric CDR-SB weighs memory deficits more heavily.
How does the CDR plus NACC FTLD do? Scores tracked reliably with diagnoses among 970 people with sporadic and familial forms of FTD who participated in the ARTFL/ LEFFTDS cohorts, now thankfully renamed ALLFTD (Miyagawa et al., 2020). Compared to the CDR-SB, which did not show deficits among people with mild behavioral or language disturbances, the CDR plus NACC FTLD reliably picked up deficits—i.e., scored above zero— among people who had been separately diagnosed with mild FTD symptoms, such as sluggishness or a slight delay in finding words. The CDR-NACC-FTLD is the primary outcome measure in Alector’s Phase 3 trial of its anti-sortilin antibody AL001.
Even as this FTLD-ified CDR makes its debut in clinical trials, ALLFTD investigators want more comprehensive, inclusive tests. One candidate is the MIR, or multidomain impairment rating, developed by Bradley Boeve at the Mayo Clinic in Rochester, Minnesota, and ALLFTD colleagues (Dec 2018 conference news). This composite tacks visuospatial, parkinsonian, and motor function domains onto the CDR plus NACC-FTLD. Casting such a wide net offers the best chance to catch and track the range of symptoms afflicting people with FTLD, Howard Rosen of University of California, San Francisco, told Alzforum.
A test like this could be especially useful in “basket trials,” which enroll participants with a common genetic mutation and/or core neuropathology even if they show different symptoms. For example, the MIR could quantify disease progression in a trial with C9ORF72 carriers who have bvFTD, ALS, or a combination of the two syndromes. In a tauopathy trial, the MIR could track symptoms of progressive supranuclear palsy, corticobasal degeneration, as well as bvFTD. Similarly, trials aimed at TDP-43 pathology might include participants with a range of clinical phenotypes that the MIR could chart.
Existing tests of executive dysfunction are also contenders for outcome measures. One, the NIH-EXAMINER, includes measures of working memory, cognitive control, word fluency, and multitasking. UCSF researchers reported that the test picks up deficits among mutation carriers who, according to their score of zero on the CDR plus NACC FTLD, are considered still presymptomatic (Staffaroni et al., 2020). However, researchers estimated that to detect a 40 percent slowing in decline on this composite, a clinical trial would need to enroll more than 2,000 participants per treatment arm. In FTD, that is unrealistic. At ICFTD, Adam Staffaroni called this number “sobering,” but said his study used no biomarkers to pick participants on the verge of symptoms, which would presumably lower the numbers of participants needed to see an effect.
Executive Inklings. Among people who are asymptomatic as per their CDR plus NACC FTLD scores, those with a pathogenic FTLD mutation (red) score lower on the NIH EXAMINER than noncarriers (black), and continue to slip over time. [Courtesy of Staffaroni et al., Alzheimer’s and Dementia, 2020.]
Staffaroni has built a Bayesian disease-progression model with data from ALLFTD and GENFI participants. It estimates an individual’s “disease age” by taking into account his or her plasma NfL, volumetric MRI, neuropsychological measures, and even CDR-NACC-FTLD score. Using such a model could help offer a patient a prognosis, and also help select the right participants for clinical trials, Staffaroni said. He is currently developing an interactive online platform that will aid in trial planning.
To run effective, sufficiently powered trials, the FTD community will need to deploy a combination of outcomes, and pool resources. One model is cooperative studies such as the DIAN-TU platform trial, which tests multiple drugs at once against a shared placebo group (Mar 2021 news).
Even a decade into GENFI, however, honing clinical outcome measures, particularly for people in the presymptomatic and earliest symptomatic stages of FTD, remains a critical challenge, said its leader, Jonathan Rohrer of University College London. “We’ve learned a lot about how MRI and fluid biomarkers change, but we still don’t have the perfect clinical outcome measure,” he conceded.—Jessica Shugart
Miyagawa T, Brushaber D, Syrjanen J, Kremers W, Fields J, Forsberg LK, Heuer HW, Knopman D, Kornak J, Boxer A, Rosen HJ, Boeve BF, Appleby B, Bordelon Y, Bove J, Brannelly P, Caso C, Coppola G, Dever R, Dheel C, Dickerson B, Dickinson S, Dominguez S, Domoto-Reilly K, Faber K, Ferrell J, Fishman A, Fong J, Foroud T, Gavrilova R, Gearhart D, Ghazanfari B, Ghoshal N, Goldman JS, Graff-Radford J, Graff-Radford N, Grant I, Grossman M, Haley D, Hsiung R, Huey E, Irwin D, Jones D, Jones L, Kantarci K, Karydas A, Kaufer D, Kerwin D, Kraft R, Kramer J, Kukull W, Litvan I, Lucente D, Lungu C, Mackenzie I, Maldonado M, Manoochehri M, McGinnis S, McKinley E, Mendez MF, Miller B, Multani N, Onyike C, Padmanabhan J, Pantelyat A, Pearlman R, Petrucelli L, Potter M, Rademakers R, Ramos EM, Rankin K, Rascovsky K, Roberson ED, Rogalski E, Sengdy P, Shaw L, Tartaglia MC, Tatton N, Taylor J, Toga A, Trojanowski JQ, Wang P, Weintraub S, Wong B, Wszolek Z.
Utility of the global CDR® plus NACC FTLD rating and development of scoring rules: Data from the ARTFL/LEFFTDS Consortium.
Alzheimers Dement. 2020 Jan;16(1):106-117.
PubMed.
Staffaroni AM, Bajorek L, Casaletto KB, Cobigo Y, Goh SM, Wolf A, Heuer HW, Elahi FM, Ljubenkov PA, Dever R, Kornak J, Appleby B, Bove J, Bordelon Y, Brannelly P, Brushaber D, Caso C, Coppola G, Dheel C, Dickerson BC, Dickinson S, Dominguez S, Domoto-Reilly K, Faber K, Ferrall J, Fields JA, Fishman A, Fong J, Foroud T, Forsberg LK, Gavrilova R, Gearhart D, Ghazanfari B, Ghoshal N, Goldman J, Graff-Radford J, Graff-Radford N, Grant I, Grossman M, Haley D, Hsiung GY, Huey ED, Irwin DJ, Jones DT, Jones L, Kantarci K, Karydas A, Kaufer DI, Kerwin DR, Knopman DS, Kraft R, Kremers WK, Kukull WA, Litvan I, Lucente D, Lungu C, Mackenzie IR, Maldonado M, Manoochehri M, McGinnis SM, McKinley E, Mendez MF, Miller BL, Multani N, Onyike C, Padmanabhan J, Pantelyat A, Pearlman R, Petrucelli L, Potter M, Rademakers R, Ramos EM, Rankin KP, Rascovsky K, Roberson ED, Rogalski E, Sengdy P, Shaw LM, Syrjanen J, Tartaglia MC, Tatton N, Taylor J, Toga A, Trojanowski JQ, Weintraub S, Wang P, Wong B, Wszolek Z, Boxer AL, Boeve BF, Kramer JH, Rosen HJ, ARTFL/LEFFTDS consortium.
Assessment of executive function declines in presymptomatic and mildly symptomatic familial frontotemporal dementia: NIH-EXAMINER as a potential clinical trial endpoint.
Alzheimers Dement. 2020 Jan;16(1):11-21.
PubMed.
Further Reading
No Available Further Reading
From Specialized to Standardized: Social-Emotional Tests for FTD
One person “checks out” from family, seeming reluctant to talk. Another talks incessantly, without saying much. One person seems unaware of the emotions of others; another recognizes emotions but doesn’t seem to care. The social and emotional manifestations of frontotemporal lobar degeneration (FTLD) are complex and manifold, like everything about these conditions. How could a clinician measure them objectively, much less build standardized tests? Still, researchers are trying, not least because sensitive clinical outcome measures are so sorely needed to find out whether an investigational drug worked in a clinical trial (see Part 7 of this series).
Because FTD is rare and complicated, large international consortia, particularly Europe’s GENFI and North America’s ALLFTD, have been key to devising such tests. Their researchers are validating older neuropsychological tests that can be broadly deployed across the globe. They are also creating new ways to probe neuronal disturbances responsible for the behavioral oddities that people with FTD, and their caregivers, deal with on a daily basis. In several recent papers, and at the International Conference for FTD, held virtually March 3–5, scientists described tests that tease out deficits in social and emotional cognition, linking them to malfunction in specific neural networks in the brain.
Social and emotional deficits form the core of some FTD syndromes, notably bvFTD and semantic PPA. At ICFTD, Katherine Rankin of the University of California, San Francisco, presented data validating a suite of tests that probe this kind of dysfunction. Rankin explained that socioemotional deficits in FTD stem from crumbling circuitry in two neural networks. She called the salience network “ground zero for bvFTD.” It picks up cues from the environment that relate to our health, safety, and survival, and provokes autonomic responses, such as quickened heart rate, elevated skin conductance, and dilated pupils. Because humans are profoundly social creatures, social cues are also critical to our well-being, hence salient, Rankin said. “If my boss rolls her eyes during my presentation, that could signal grave consequences for my professional future,” Rankin said.
The semantic appraisal network is also under attack in FTD. “The SAN is a superhighway that connects valence to semantics. It supports socioemotional processing in many ways,” said Rankin. Essentially, the SAN connects semantic information to the appropriate emotional response. In the eye-rolling example, the SAN would help the presenter decide whether to feel threatened by the eye roll, or perhaps to laugh instead if the eye-rolling referred to a shared joke between friends.
While it’s clear these networks are disrupted in FTD, Rankin said that testing the degree of that with neuropsychological tests is a tall order, because it’s difficult to “fake salience” in a testing scenario. One way to approximate this is by measuring autonomic, physiological responses, such as skin conductance or pulse. A slew of candidate tests do that, but Rankin emphasized that the field urgently awaits validated neuropsychological tests, which require no special equipment and can be employed across centers and cultures.
Rankin and colleagues have been validating four socioemotional tests in NACC’s FTLD module. Most rely on an informant—a caregiver or clinician—to gauge social impairment in the person with FTD. Rankin’s personal favorite is the revised self-monitoring scale (RSMS), which gauges a person’s ability to react to subtle social cues. The test asks a close relative or friend of the patient to rank his or her social awareness and responsiveness, as gauged by how true 13 statements are. For example, one is “In conversations, the subject is sensitive to even the slightest change in the facial expression of the other person he/she is conversing with.”
Previously, the researchers had reported from work with 168 people that scores on this test distinguished those with FTD from healthy controls and, perhaps more importantly, could differentiate among FTD syndromes (Toller et al., 2018). As expected, people with bvFTD performed worst, followed by those with semantic primary progressive aphasia, Rankin said. A more recent study in the ALLFTD cohort, of 475 participants including presymptomatic mutation carriers, found changes in the RSMS prior to symptom onset that worsened with disease progression (Toller et al., 2020). In a subset of participants who returned for more than one visit, more rapid annual decline on the RSMS was significantly associated with faster shrinkage of regions belonging to the SN and SAN, such as the right anterior insula, dorsal anterior cingulate, and orbitofrontal cortex.
Messy but Trendy. Scores on the RSMS, a test of a person’s ability to respond to social cues, trended steadily downward as disease progressed among mutation carriers with bvFTD. [Courtesy of Toller et al., Neurology, 2020.]
The interpersonal reactivity index is another test that relies on information from a caregiver or close friend. The IRI goes a step further than the RSMS. It assesses a person’s ability to put him- or herself in another’s shoes, make an effort to understand another person’s perspective, and exhibit feelings of empathic concern when they see someone in need. Essentially, it’s a gauge of empathy. At ICFTD, Rankin reported that in the ALLFTD cohort, scores on this test tracked with disease severity as gauged by the CDR plus NACC FTLD test battery (see Part 1 of this series). For some mutation carriers, a score of zero on the CDR NACC FTLD indicated they were asymptomatic; however, their caregivers begged to differ, reporting a dearth of empathy in their partners. Similar to the RSMS, people with bvFTD had the strongest deficits on the IRI, followed by people with semantic variant of PPA. Here, too, lower scores were tied to shrinkage in key hubs of the salience and semantic appraisal networks.
Two other tests—the social behavior observer checklist (SBOCL) and the Social Norms Questionnaire (SNQ)—are also proving useful in picking up deterioration of the salience and semantic appraisal networks in ALLFTD, Rankin reported at ICFTD. Rankin and colleagues developed the SBOCL more than a decade ago to help clinicians distinguish between people with bvFTD and those with psychiatric disorders. The SBOCL is completed by a clinician, rather than a close friend or caregiver of the person with FTD (Rankin et al., 2008). After spending time with a patient, a clinician responds to questions such as “Was [the subject] overly disclosing or inappropriately familiar?” The SNQ determines the degree to which a person understands implicit but widely accepted social boundaries in American culture. For this test, the participant answers questions such as, “Would it be socially acceptable to laugh when someone else trips and falls?” Together with the RSMS and IRI, these tests of social cognition are part of the NACC-FTLD module and Gefen et al., 2020). Rankin and colleagues are about to publish a study that weaves in data from more than 1,300 people who were evaluated using this module in the NACC database. At ICFTD, Rankin reported that all four measures distinguished between people with different forms of FTD.
Most of these tests need informants. Rankin and colleagues are also devising tests that require only people with FTD, or presymptomatic mutation carriers, themselves. For example, the Dynamic Affect Recognition Test (DART) designed at UCSF plays a video of someone talking on the phone to participants, and asks them to gauge the speaker’s emotions by looking at his or her facial expressions, which change over the course of the video. The researchers are currently testing a tablet version in healthy volunteers.
These tests are not just a hodgepodge of quirky tests that work only at one center or another. The FTD field is now equipped with a growing toolkit of socioemotional measures that have been validated in large international cohorts, Rankin said. Even so, more are still needed, especially tests that work across languages and cultures. She called for investigators to keep publishing and validating neuropsych tests for FTD, even as they develop their own specialized tests to address their favorite question at their own center.
Tests of social cognition are also moving up the ranks of GENFI, the multicenter observational study of familial FTD with centers in Europe and Canada. Jason Warren of University College London heads the brain-behavior group there. Warren wants to understand how behavioral symptoms that patients and their families report to their neurologists relate to brain changes. “We want to deconstruct these symptoms into their building blocks, and find out where the mischief is in the brain,” he said. Over the past few years, Warren’s group has devised a slew of tests that use stimuli such as sound, sarcasm, humor, or body awareness, and physiological responses like pupil dilation or heart rate, to identify specific deficits in people with FTD and to zero in on their neurological bases (Nov 2014 conference news).
They have begun to roll out these tests within the larger GENFI cohort. One example is a modified version of the Camel and Cactus test, a measure of semantic knowledge. It asks participants to match pictures or words based on semantic relationships, i.e. “camel” goes with “cactus,” not “tree,” “sunflower,” or “rose.” The researchers adapted the test for use in multiple languages and cultures. Among 664 GENFI participants, symptomatic carriers of C9ORF72, GRN, or MAPT mutations performed worse than noncarriers, and deficits even emerged among presymptomatic carriers of C9ORF72 or MAPT mutations (Moore et al., 2020). The semantic deficits tracked with different atrophy patterns among the different groups of mutation carriers, correlating with atrophy in the temporal lobe in MAPT carriers, in both the temporal and frontal lobes in C9ORF72 carriers, and with shrinkage in the left frontal lobe in GRN carriers.
Two other tests helped GENFI detect early social and emotional deficits among mutation carriers. In one, participants were shown faces and asked to name the emotions on them. In the other, they were shown a series of short cartoon stories, and asked to identify those that contained a social inconvenience, or faux pas (Russell et al., 2020). Compared to controls, symptomatic people with familial FTD fared much worse on both tests. Even presymptomatic deficits cropped up in C9ORF72 carriers, who had difficulty recognizing fear and sadness, but not other emotions or faux pas. Importantly, poor performance on these tests correlated with atrophy in regions of the brain known to facilitate social cognition.
A new study in GENFI’s Dutch cohort reached similar conclusions. It used a more sensitive gauge of emotion recognition that asked participants to name emotions on faces showing different intensities of emotion (Jiskoot et al., 2021). In this computer-based test, most people more accurately named an emotion as its intensity increased. However, 32 people with Alzheimer’s disease and 32 with bvFTD scored lower across most emotions displayed than did 47 presymptomatic FTD mutation carriers and 49 healthy controls. People with bvFTD performed markedly worse than those with AD in deciphering anger and happiness. Presymptomatic mutation carriers mostly performed at the level of controls, except for C9ORF72 carriers, who had a hard time identifying mild disgust.
Name the Emotion. Shown images of faces displaying different intensities of one of six emotions, people with bvFTD and people with AD had a hard time labeling the emotion, especially those at low intensity. People with bvFTD scored low, especially on anger, happiness, disgust. [Courtesy of Jiskoot et al., Journal of Neurology, 2021.]
For his part, Warren is exploring better ways to tease out emotional problems among the London cohort of FTD families. For example, he noted that traditional tests of facial emotion recognition rely on complex instructions and require high levels of working memory on the part of the patient, which can confound results. To avoid this, he tracked people’s gazes to gauge emotional recognition (Russell et al., 2021). Participants were given no instructions except to observe images. As they looked at faces or, in a more difficult version of the task, sets of eyes displaying different emotions, a scanner tracked the participants’ own eye movements. Later, when shown a word fitting one of the emotions, i.e., “happy,” controls tended to rest their eyes on the face that matched that emotion, but people with bvFTD rarely zeroed in on any particular face, suggesting they did not recognize the emotion. Worse performance in the bvFTD group correlated with atrophy in the right ventromedial prefrontal and orbitofrontal cortices, brain regions previously implicated in social cognition.
Eye of the Beholder. Healthy controls spent more time looking at faces (top) or sets of eyes (bottom) that matched the displayed emotion. People with bvFTD did not focus on any one face. The color bar reflects time spent gazing at a particular area. [Courtesy of Russell et al., Alzheimer’s Research & Therapy, 2021.]
At ICFTD, Fiona Kumfor of the University of Sydney reported that people with bvFTD focus on different regions of a person’s face. Also using eye tracking, Kumfor and colleagues found that, when presented with a face showing an emotion, people with bvFTD spent more time focused on the eyes than did healthy controls. They titled their paper “Looking but not seeing” (Hutchings et al., 2018). This was counterintuitive, Kumfor said, as people with other behavioral disorders, such as autism, tend to avoid looking at eyes. Alas, their extended focus on eyes did not help people with bvFTD recognize emotions. Kumfor’s previous studies had also used physiological measures such as skin conductance and smiling/frowning measurements to reveal that people with bvFTD do not experience emotional scenes in the same way healthy people do (Kumfor et al., 2019).
That people with bvFTD would home in on eyes dovetails with findings from what Kumfor and colleagues call “a truly social approach” to measuring social cognition, i.e., just watching people converse (Visser et al., 2020). The investigators observed a 10-minute video of participants chatting with a clinician. They took note of socially engaged behaviors such as nods, smiles, and gestures, and disengaged ones, like avoiding eye contact, self-grooming, or interrupting. Compared to 20 participants with semantic dementia and 20 with Alzheimer’s, 20 people with bvFTD tended to nod less but also look away less. In fact, they tended to stare for long periods of time, a behavior that people find unsettling in social situations, Kumfor said. People with SD, on the other hand, gestured more often while talking, perhaps reflecting their reliance on nonverbal communication.—Jessica Shugart
Moore K, Convery R, Bocchetta M, Neason M, Cash DM, Greaves C, Russell LL, Clarke MT, Peakman G, van Swieten J, Jiskoot L, Moreno F, Barandiaran M, Sanchez-Valle R, Borroni B, Laforce R Jr, Doré MC, Masellis M, Tartaglia MC, Graff C, Galimberti D, Rowe JB, Finger E, Synofzik M, Karnath HO, Vandenberghe R, de Mendonça A, Maruta C, Tagliavini F, Santana I, Ducharme S, Butler C, Gerhard A, Levin J, Danek A, Otto M, Warren JD, Rohrer JD, Genetic FTD Initiative, GENFI*.
A modified Camel and Cactus Test detects presymptomatic semantic impairment in genetic frontotemporal dementia within the GENFI cohort.
Appl Neuropsychol Adult. 2020 Feb 5;:1-8.
PubMed.
Russell LL, Greaves CV, Bocchetta M, Nicholas J, Convery RS, Moore K, Cash DM, van Swieten J, Jiskoot L, Moreno F, Sanchez-Valle R, Borroni B, Laforce R Jr, Masellis M, Tartaglia MC, Graff C, Rotondo E, Galimberti D, Rowe JB, Finger E, Synofzik M, Vandenberghe R, de Mendonça A, Tagliavini F, Santana I, Ducharme S, Butler C, Gerhard A, Levin J, Danek A, Otto M, Warren JD, Rohrer JD, Genetic FTD Initiative, GENFI.
Social cognition impairment in genetic frontotemporal dementia within the GENFI cohort.
Cortex. 2020 Dec;133:384-398. Epub 2020 Sep 26
PubMed.
Convery RS, Bocchetta M, Greaves CV, Moore KM, Cash DM, Van Swieten J, Moreno F, Sánchez-Valle R, Borroni B, Laforce R Jr, Masellis M, Tartaglia MC, Graff C, Galimberti D, Rowe JB, Finger E, Synofzik M, Vandenberghe R, de Mendonca A, Tagliavini F, Santana I, Ducharme S, Butler C, Gerhard A, Levin J, Danek A, Otto M, Warren JD, Rohrer JD, Genetic FTD Initiative (GENFI).
Abnormal pain perception is associated with thalamo-cortico-striatal atrophy in C9orf72 expansion carriers in the GENFI cohort.
J Neurol Neurosurg Psychiatry. 2020 Dec;91(12):1325-1328. Epub 2020 Aug 5
PubMed.
Requena-Komuro MC, Marshall CR, Bond RL, Russell LL, Greaves C, Moore KM, Agustus JL, Benhamou E, Sivasathiaseelan H, Hardy CJ, Rohrer JD, Warren JD.
Altered Time Awareness in Dementia.
Front Neurol. 2020;11:291. Epub 2020 Apr 21
PubMed.
Cohorts Band Together to Get Global FTD Trials Going
Compared to Alzheimer’s disease, frontotemporal lobar degeneration is far rarer and more diverse in terms of its clinical manifestations and their underlying genetic and neuropathological causes. This makes the illness maddeningly difficult to understand at every level, let alone target with disease-modifying therapies. On the former front, scientists have made real progress charting FTLD’s complexity through cohort studies. Europe’s GENFI and North America’s ALLFTD have dutifully tracked the variable trajectories of the disease and hunted for fluid, neuroimaging, and cognitive markers of its progression (Parts 1, 2, 3, 4, and 5 of this series). Now, the field is poised to put that observational rubber to the road. The FTD Prevention Initiative is the über-consortium that unites multicenter cohort studies across the globe. The FPI aims to harmonize FTD data from around the world, work with industry to design and execute international clinical trials, and create an international registry of participants keen to join. As its name implies, another goal is to enroll presymptomatic mutation carriers in trials, i.e., eventually prevent FTD.
In an FPI paper, and at the International Conference for FTD, held virtually March 3–5, researchers reported the fruits of their initial efforts to pull together data from the cohorts under the FPI umbrella. In particular, the meeting featured a session dedicated to a new Latin American project that is poised to expand knowledge about all aspects of FTD, and will join FPI.
Launched in 2019, FPI is led jointly by Jonathan Rohrer of University College London, who heads GENFI, and Adam Boxer of the University of California, San Francisco, who co-leads ALLFTD (Rohrer and Boxer, 2021). Other observational studies within FPI are Australia’s Dominantly Inherited Non-Alzheimer Dementias study (DINAD), New Zealand’s FTD Genetic Study (FTDGeNZ) and, importantly, South America’s Research Dementia Latin America (ReDLaT) study. Patient-advocacy groups and FTD research foundations have also signed on, including the Association for Frontotemporal Degeneration, the Bluefield Project to Cure FTD, and the FTD Disorders Registry.
The Struggle for Power
FPI addresses a tall hurdle standing in the way of effective clinical trials for FTD, AFTD’s Susan Dickinson told Alzforum. It’s how to achieve power in a rare, heterogeneous disease. Only collaborative, international clinical trials will be able to recruit enough participants to detect a statistically significant effect. “The more we can do to pool data and work together, the easier it will be to make these trials a reality,” Dickinson said, adding, “The small number of available patients will be a defining feature of how we approach these trials, no question.”
FPI’s first order of business is to harmonize data from longitudinal cohort studies across countries. So far, FPI has one paper out that weaves in data from GENFI, ALLFTD, and DINAD. Published just as the COVID-19 pandemic started its march across the globe, the study addressed basic parameters of familial FTD, comparing age at disease onset and at death among people who carried a pathogenic mutation in C9ORF72, GRN, or MAPT (Moore et al., 2020).
The largest study of familial FTD to date, it reports data from 3,403 people from 1,492 families. Behavioral variant FTD was the most common clinical syndrome, but people with primary progressive aphasias, amyotrophic lateral sclerosis, corticobasal degeneration, and progressive supranuclear palsy were also included. Of the participants, 1,433 had a C9ORF72 hexanucleotide expansion, 1,179 had a GRN mutation, and 791 had a MAPT mutation. The frequency of mutations was somewhat different in different parts of the world. For example, GRN mutations were most prevalent in Italy and Spain, while MAPT mutations were predominant in the Netherlands and on the west coast of the United States (see below).
Global Tally. The number of FPI participants who carry a mutation in C9ORF72, GRN, or MAPT is shown for each region included. Countries shaded in dark blue participated in the study; individual centers in red. [Courtesy of Moore et al., Lancet Neurology, 2020.]
How did timing and severity of the disease differ between mutation carriers? MAPT mutation carriers showed symptoms first, at an average age of 49, followed by C9ORF72 carriers at 58, and GRN carriers at 61. MAPT carriers lived for an average of 9.3 years after diagnosis, while C9ORF72 and GRN carriers lived for an average of 6.4 and 7.1 years, respectively. Within each mutation group, age at onset varied markedly, ranging from the 20s to the 90s in the GRN and C9ORF72 groups, and from age 17 to the 80s in the MAPT group.
When FTD Starts. Age at symptom onset and death, charted for more than 3,400 carriers of autosomal-dominant mutations in GRN, MAPT, and C9ORF72. For each causative gene, the age span is enormous. [Courtesy of Moore et al., Lancet Neurology, 2020.]
Across the cohort, men and women were of similar age at symptom onset. However, symptoms cropped up later in women than men in the GRN and C9ORF72 groups.
Within families burdened by FTD across generations, the scientists found, alarmingly perhaps, that symptoms emerged a few years earlier with each successive generation across all mutation groups. The strength of the association between an individual’s age at onset with that of their parent’s varied among the groups, with MAPT mutations having a strong tie, whereas in C9ORF72 or GRN carriers, the age at which their affected parent had gotten sick poorly predicted when the next generation would.
With the basic parameters of FTD now laid down across mutations, FPI’s next data merger will compare measurements of plasma NfL, the most advanced FTD biomarker to date, across the initiative’s cohorts. An upcoming paper shows remarkable agreement among the cohorts in the relationship between plasma NfL and symptom onset, Boxer said, but its authors declined to say more about it while the manuscript is being reviewed.
Latin American Cohort Joins the Fight
To date, most FPI participants are of European descent. This is about to change, as the Multi Partner Consortium to Expand Research in Latin America (ReDLaT) joins the effort. This cohort includes people with FTD, but also AD and healthy controls, in Argentina, Brazil, Chile, Colombia, Mexico, and Peru. Funded by the National Institutes of Health, the Alzheimer’s Association, and others, the consortium is jointly led by Agustín Ibañez at Universidad de San Andrés, Buenos Aires, and Bruce Miller at the University of California, San Francisco.
ReDLaT aims to enroll 4,500 participants. In accordance with the prevalence of different dementias, the majority of participants will be healthy controls or people with AD, followed by people with FTD. The study will track them with clinical, neuroimaging, and fluid biomarker measurements, applying protocols already in use within the ALLFTD cohort. The COVID-19 pandemic slowed down this project, as it did clinical research everywhere, but ReDLaT still managed to get 11, soon to be 12, study sites up and running, Ibañez said at ICFTD.
ReDLaT aims to establish the genetic basis of AD and FTD in diverse Latin American cohorts. Jennifer Yokoyama of UCSF heads ReDLaT’s genetics effort. She explained that the diversity of genetic backgrounds in South American populations—which include indigenous peoples, people of African descent, Europeans and, of course, mixtures of all these—is sure to increase the number of genetic variants linked to FTD. At ICFTD, Yokoyama offered a sneak peek of preliminary genetic data collected in ReDLaT so far, reporting unexpected pathogenic variants, such as in LRRK2 and Notch3, in people with AD. As genomic studies expand in the cohort, the scientists can test how polygenic risk scores (PRS), which so far have only been validated in non-Hispanic white populations, perform in Hispanic people, as well.
Aside from FPI, ReDLaT also has its own goals in advancing the understanding of dementia syndromes in Latin America. One aim is to assess how socioeconomic status, low access to formal education and healthcare, and exposure to political and gender violence—all factors with strong sway on the overall health of populations in Latin America—might influence manifestations of FTD there. At ICFTD, ReDLaT researchers Andrea Slachevsky of the University of Chile and Stefanie Piña Escudero of the Global Brain Health Institute reported on how measures of these so-called social determinants of health are being incorporated into clinical assessments of people in the ReDLaT cohort. Separately, they hope to educate physicians and the public about FTD, which is still a relatively unknown disease in South America, Ibañez said.—Jessica Shugart
Moore KM, Nicholas J, Grossman M, McMillan CT, Irwin DJ, Massimo L, Van Deerlin VM, Warren JD, Fox NC, Rossor MN, Mead S, Bocchetta M, Boeve BF, Knopman DS, Graff-Radford NR, Forsberg LK, Rademakers R, Wszolek ZK, van Swieten JC, Jiskoot LC, Meeter LH, Dopper EG, Papma JM, Snowden JS, Saxon J, Jones M, Pickering-Brown S, Le Ber I, Camuzat A, Brice A, Caroppo P, Ghidoni R, Pievani M, Benussi L, Binetti G, Dickerson BC, Lucente D, Krivensky S, Graff C, Öijerstedt L, Fallström M, Thonberg H, Ghoshal N, Morris JC, Borroni B, Benussi A, Padovani A, Galimberti D, Scarpini E, Fumagalli GG, Mackenzie IR, Hsiung GR, Sengdy P, Boxer AL, Rosen H, Taylor JB, Synofzik M, Wilke C, Sulzer P, Hodges JR, Halliday G, Kwok J, Sanchez-Valle R, Lladó A, Borrego-Ecija S, Santana I, Almeida MR, Tábuas-Pereira M, Moreno F, Barandiaran M, Indakoetxea B, Levin J, Danek A, Rowe JB, Cope TE, Otto M, Anderl-Straub S, de Mendonça A, Maruta C, Masellis M, Black SE, Couratier P, Lautrette G, Huey ED, Sorbi S, Nacmias B, Laforce R Jr, Tremblay ML, Vandenberghe R, Damme PV, Rogalski EJ, Weintraub S, Gerhard A, Onyike CU, Ducharme S, Papageorgiou SG, Ng AS, Brodtmann A, Finger E, Guerreiro R, Bras J, Rohrer JD, FTD Prevention Initiative.
Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study.
Lancet Neurol. 2020 Feb;19(2):145-156. Epub 2019 Dec 3
PubMed.
FPI’s raison d’etre is to get effective, international clinical trials on the road (see Part 6 of this series). At ICFTD, FPI’s co-leader, Adam Boxer of the University of California, San Francisco, summarized what trials exist already, and offered a glimpse of the future.
Currently, the field lacks biomarkers for the underlying neuropathology of FTD, so there is no concrete way to know whether tau fibrils or TDP-43 inclusions lurk within the brain of a patient who has no known autosomal-dominant mutation. Hence, present-day FTD trials are limited to carriers of pathogenic C9ORF72, GRN, or MAPT mutations, and to people whose clinical syndromes are known to be tauopathies, such as progressive supranuclear palsy.
Trials for carriers of GRN mutations are the farthest along. At centers that are part of FPI, Alector is conducting Phase 2 and Phase 3 trials evaluating AL001, an anti-sortilin antibody meant to slow progranulin’s degradation. Early trials of AL001 indicated that it restores CSF progranulin levels in mutation carriers. The Phase 3 trial is the first in the field to include presymptomatic mutation carriers. They can enroll if their plasma NfL is elevated, suggesting they are nearing symptom onset. The trial aims for 180 participants total, and also includes symptomatic carriers. It uses the CDR-NACC-FTLD as a clinical primary endpoint, and is slated to run through 2023.
Two companies—Prevail Therapeutics (recently acquired by Eli Lilly & Company) and Passage Bio—are pursuing a gene-therapy approach. Both are hoping to replenish progranulin levels by injecting an adeno-associated virus bearing the progranulin gene directly into the cisterna magna of GRN mutation carriers with FTD. Prevail’s trial of PR006 started in July 2020, while Passage Bio’s trial of PBFT02 is slated to start this month.
Julio Rojas of the University of California, San Francisco, said the combination of sensitive disease biomarkers (CSF progranulin, plasma NfL, and others) and progranulin-targeted treatment in trials bodes well for this form of familial FTD. “It’s likely we’ll see a treatment for GRN mutation carriers with FTD before we see one for AD,” Rojas said.
Carriers of hexanucleotide expansions in the C9ORF72 gene are also being included within the progranulin umbrella. Owing to the lysosomal dysfunction wrought in both familial forms of the disease, C9ORF72 carriers are included in Alector’s Phase 2 study of AL001.
Other trials are targeting the C9ORF72 mutation specifically. They enroll people with ALS and/or FTD. The furthest along is Ionis/Biogen’s antisense oligonucleotide BIIB078, in Phase 1. The multiple-ascending-dose trial includes 114 people with ALS; results are expected later this year. Participants in the randomized portion of the trial are being enrolled in a two-year, open-label extension. Wave Life Sciences, Cambridge, Massachusetts, is planning to start a Phase 1 trial of its C9-ASO, called WVE-004, in people with ALS and FTD due to C9ORF72 expansion this year (see press release).
Yet another C9-ASO, called afinersen, was designed in Robert Brown’s group at the University of Massachusetts in Worcester. At ICFTD, Boxer showed data from a man with ALS who was treated with afinersen for over a year. During this time, his CSF levels of poly-GP, a dipeptide translated from transcripts of the hexanucleotide expansion, plummeted, and his disease stayed stable during treatment.
The diabetes drug metformin is being tested at the University of Florida, Gainesville, in a Phase 1 clinical trial for C9ORF72 hexanucleotide expansion carriers with ALS or FTD. Besides its better-known effects on glucose levels and insulin sensitivity, metformin has been reported to squelch repeat-associated non-AUG (RAN) translation, the mechanism responsible for translating toxic dipeptides from the C9 repeats (Zu et al., 2020). This open-label study includes 18 participants, and is expected to finish in August 2022.
What about trials for FTLD-tau? So far, most trials have focused on people with clinical syndromes, such as progressive supranuclear palsy, which is known to have underlying tau pathology, but not necessarily a pathogenic tau mutation. Two different anti-tau monoclonal antibodies failed in trials for PSP last year (Jul 2019 news; Dec 2019 news). This prompted cancellation of a “basket trial” that had also included people with non-fluent primary progressive aphasia, corticobasal syndrome, and MAPT mutation carriers with FTD.
Why did these trials fail? A definitive answer is not in, but Boxer noted that CSF studies from the trials indicated that both antibodies, which target tau’s N-terminus, did not rid the brain of all forms of tau pathology. Boxer said the field eagerly awaits results of Biogen’s trial of BIIB080, a tau-targeting ASO in people with mild AD, which could provide proof-of-concept data for primary tauopathies. That Phase 2 trial will finish in May of 2022.
One hurdle to treating primary tauopathies is the lack of fluid biomarkers or PET imaging tracers for many non-AD forms of tau. While some tracers have been found to bind to the 4R-tauopathies PSP and CBD, so far none reliably tag tau in people with bvFTD or PPA, who could have underlying tau or TDP-43 pathology (Jul 2020 news). Once the field develops a marker for either one, people with sporadic FTD will be able to join clinical trials targeting their specific pathology.
MAPT mutations are the rarest cause of familial FTD, i.e., patients with these mutations are scarce. To ensure that trials in this far-flung group of patients are run with the most power possible, the FPI is working to coordinate international platform trials for them, which will test multiple therapies simultaneously against a shared placebo group, Boxer said.—Jessica Shugart


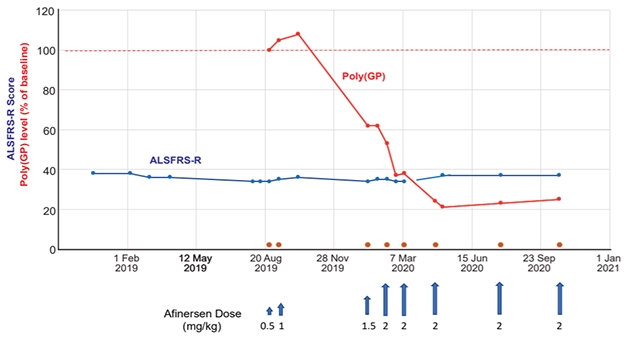


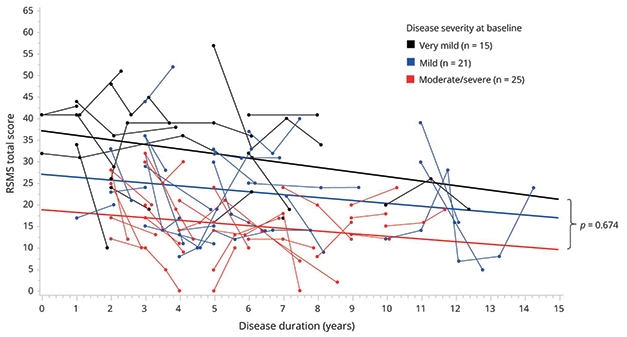
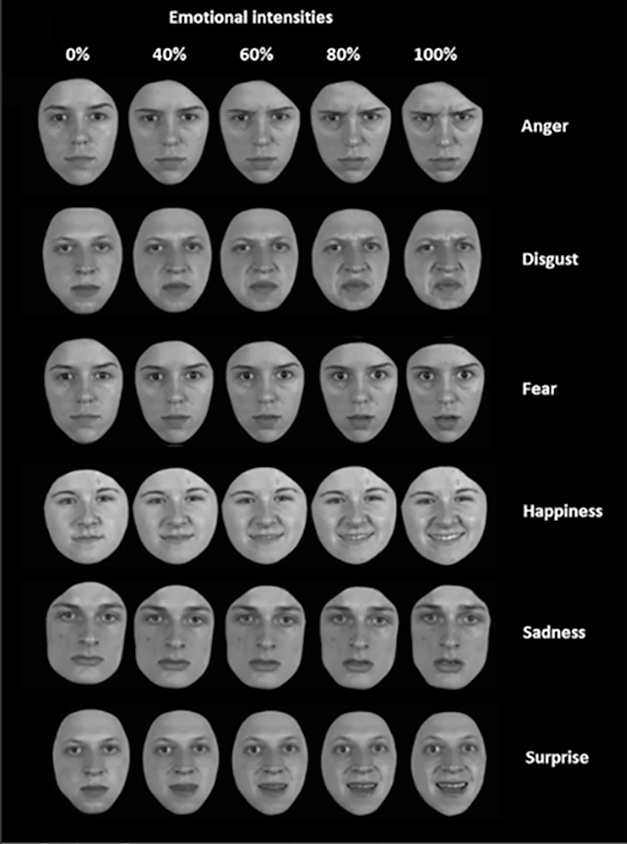
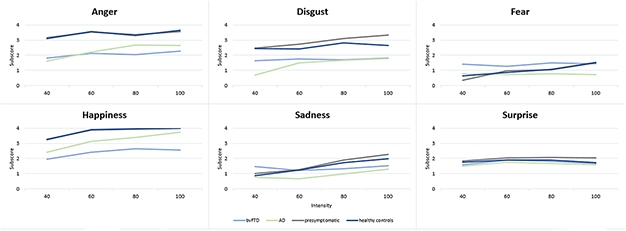



Comments
No Available Comments
Make a Comment
To make a comment you must login or register.