Cut Loose, Soluble TREM2 Beckons Microglia to Mop Up Plaques
Quick Links
As if the biological functions of membrane-bound TREM2 weren’t complicated enough, researchers also have to contend with the actions of its freewheeling counterpart, soluble TREM2. In the March 25 Nature Communications, researchers led by Guojun Bu at the Mayo Clinic in Jacksonville, Florida, and Xiao-Fen Chen of Xiamen University in China reported that injecting soluble TREM2 (sTREM2) into the brains of AD model mice activated microglia and rallied them to encircle and engulf plaques. The soluble fragment, which is shed from the microglial membrane, also corrected synaptic and memory deficits in the animals, the researchers claim. They have yet to nail down the enigmatic receptor for sTREM2, but zeroed in on microglia as the cells that express it.
- Upping sTREM2 boosted plaque clearance in 5xFAD mice.
- The fragment recruited microglia to plaques, and enhanced their phagocytosis.
- It restored synaptic function and rescued memory loss.
“Studies like this are crucial to understand the functions of both full-length receptor and its cleaved ectodomain, and are extremely instrumental in aiding design strategies to therapeutically target TREM2,” commented Gernot Kleinberger of ISAR Bioscience in Planegg, Germany.
Since the discovery of TREM2 as a major AD risk factor (see Research Timeline, 2012), enormous strides have been made in unraveling its complex function in the brain. In a nutshell, stimulation of the receptor by ligands including phospholipids, Aβ, and ApoE, summon microglia to clean up debris, including plaques or degenerating cells (Feb 2015 news; Oct 2015 news). In some animal models of amyloidosis and tauopathy, signaling through Trem2 can switch from beneficial to detrimental as disease progresses (Jul 2018 news).
Making matters more complex, ADAM proteases cleave the receptor, shedding the extracellular sTREM2 domain into the interstitial environs. This soluble bit has been spotted mingling with neurons around Aβ plaques in mice (Jan 2018 news). In people, sTREM2 spikes in the cerebrospinal fluid in the early stages of AD (Jan 2016 news).
In 2017, Bu and colleagues reported that sTREM2 bolstered microglial survival and upped pro-inflammatory cytokines (Feb 2017 news).
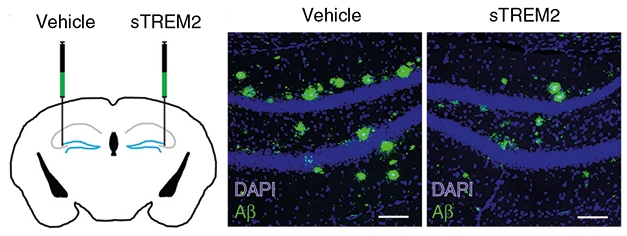
sTREM2 Lessens Plaques. Plaque load dropped by a third in the hemisphere of hippocampus injected with sTREM2 (right). [Courtesy of Zhong et al., Nature Communications, 2019.]
In their new paper, the researchers extended their investigation of sTREM2 to the 5xFAD model. First authors Li Zhong and Ying Xu, both at Xiamen University, and colleagues injected a recombinant form of sTREM2 into the right hippocampus of plaque-ridden, 7-month-old mice. They injected saline into the left hippocampus. A week later, they found that the plaque burden in the right hippocampus was a third lower than in the left. The sTREM2 injection led to the greatest reduction in large plaques, which dropped by more than half, but also reduced the total area of dystrophic neurites.
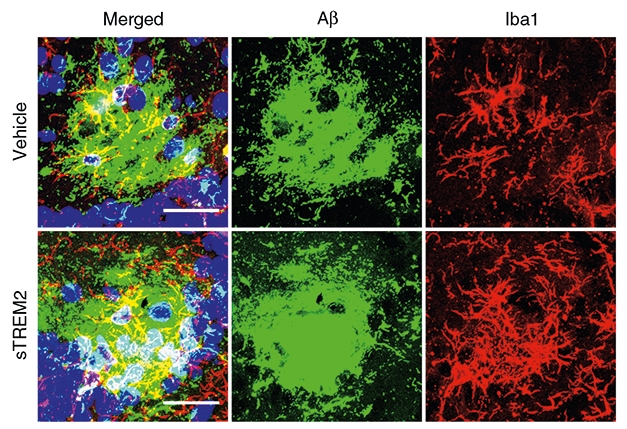
Attack the Plaque. More microglia (red) swarmed Aβ plaques (green) in mice injected with sTREM2 (bottom panels). [Courtesy of Zhong et al., 2019]
How did sTREM2 vanquish plaques? As suspected, the receptor riled up microglia. The glial cells proliferated in the hippocampus in the days following sTREM2 injection, and pumped out pro-inflammatory cytokines, including IL-1β and TNF. The microglia also surrounded plaques and expressed the phagocytic marker CD68. In a dish, primary microglia migrated toward sTREM2, and treatment with sTREM2 triggered microglia to gobble up fluorescently labeled Aβ42 via phagocytosis. Overall, the data suggested that sTREM2 both attracted microglia and stoked their appetite for Aβ.
Curiously, the effects of sTREM2 appeared to go beyond plaque clean-up. In hippocampal slice cultures from 5xFAD mice, it rescued deficits in long-term potentiation of Schaffer collaterals, a major hippocampal circuit. Soluble TREM2 also boosted expression of synaptic receptors in both the pre- and postsynapses.
Notably, when the researchers treated 5xFAD mice with PLX3397, a CSF1R receptor inhibitor that depletes the brain of microglia, sTREM2 treatment no longer boosted plaque clearance, microglial activation, or synaptic plasticity. This suggested that the beneficial effects of sTREM2—even on synaptic signaling—are mediated through microglia.
If it boosts plasticity, would sTREM2 rescue memory problems? To find out, the researchers expressed it long-term. They injected neonatal wild-type and 5xFAD mice with an adeno-associated virus carrying an sTREM2 construct, then put their spatial memory to the test in the Morris water maze six months later. Among mice expressing a control virus, wild-type mice learned the location of a submerged platform faster than did 5xFAD mice, but sTREM2 overexpression rescued this deficit in the 5xFAD animals. The AAV-TREM2 dramatically reduced plaque load, recruited microglia to plaques, and boosted synaptic function.
Bu proposed that in a physiological setting, sTREM2 amplifies and maintains the protective responses instigated by full-length TREM2. By the early symptomatic stage of AD, Aβ plaques are rampant, tau pathology has spread into the cortex, and neurons are degenerating. These injury signals might exacerbate the shedding of sTREM2 from microglia surrounding plaques and damaged cells, Bu said. In addition to binding many of the same ligands its full-length counterpart latches onto, sTREM2 likely binds an unknown receptor on microglia as well, recruiting more of the cells and potentiating their activation, Bu added. He hypothesized that the subsequent drop in sTREM2 in the CSF reflects the die-off of microglia.
“I find it very interesting that sTREM2 can promote functions that we thought are associated with membrane bound full-length TREM2,” commented Christian Haass of the German Center for Neurodegenerative Diseases in Munich. “This indeed makes therapeutic modulation of TREM2 even more tricky.” Haass added that identification of the sTREM2 receptor could shed more light on the complicated functions of microglia.
Kleinberger added that it would have been interesting to test whether mutant forms of sTREM2, such as the AD-linked R47H variant, would generate a similar loss-of-function phenotype as described for the full-length mutant receptor. The full-length receptor has so far been associated with beneficial functions such as microglial phagocytosis (Kleinberger et al., 2014). However, one study indicated that too much sTREM2 could be harmful: People harboring the H157Y mutation, which enhances sTREM shedding, had an increased risk of AD (Jiang et al. 2016).—Jessica Shugart
References
News Citations
- TREM2 Buoys Microglial Disaster Relief Efforts in AD and Stroke
- Alzheimer’s Risk Genes Interact in Immune Cells
- TREM2: Diehard Microglial Supporter, Consequences Be DAMed
- New Mouse Models Reveal Unexpected Property of TREM2
- TREM2 Goes Up in Spinal Fluid in Early Alzheimer’s
- Does Soluble TREM2 Rile Up Microglia?
Research Models Citations
Paper Citations
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
- Jiang T, Tan L, Chen Q, Tan MS, Zhou JS, Zhu XC, Lu H, Wang HF, Zhang YD, Yu JT. A rare coding variant in TREM2 increases risk for Alzheimer's disease in Han Chinese. Neurobiol Aging. 2016 Jun;42:217.e1-3. Epub 2016 Mar 3 PubMed.
Other Citations
Further Reading
Primary Papers
- Zhong L, Xu Y, Zhuo R, Wang T, Wang K, Huang R, Wang D, Gao Y, Zhu Y, Sheng X, Chen K, Wang N, Zhu L, Can D, Marten Y, Shinohara M, Liu CC, Du D, Sun H, Wen L, Xu H, Bu G, Chen XF. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer's disease model. Nat Commun. 2019 Mar 25;10(1):1365. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Biomedizinisches Centrum (BMC), Biochemie & Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
I find it very interesting that sTREM2 can promote functions that we thought are associated with membrane bound full-length TREM2. This indeed makes therapeutic modulation of TREM2 even more tricky, since we now have not only to consider “overactivation” of microglia but maybe even reduced sTREM2 function. Combined with increased ApoE expression by microglia in disease-associated states, modulation of microglia activity is a huge challenge. Identification of the sTREM2 receptor on microglia may now shed more light on the more and more complicated life of these cells.
ISAR Bioscience
TREM2 is a type-I transmembrane protein that can undergo proteolytic shedding to release its Ig-like domain as a soluble protein, termed sTREM2, into the extracellular space. It can be readily detected in biological fluids, including the cerebrospinal fluid (CSF). Shedding is mediated by ADAM proteases and occurs at a defined amino acid position after histidine 157 (Schlepckow et al., 2017; Thornton et al., 2017). Additionally, alternative splice variants have been identified in human brain that code for sTREM2 with a slightly altered C-terminus (Jin et al., 2014).
So far we know that in mouse models of amyloidosis sTREM2 levels increase dramatically (Brendel et al., 2017), while in humans the increase is moderate with a peak around the time of symptom onset with significant increased TREM2 levels starting from five years before until approximately five years after expected years of symptom onset (Suarez-Calvet et al., 2016). So far the physiological and pathophysiological role of sTREM2 remains largely understudied.
With the present study the authors follow up on a first study where they reported a positive effect of sTREM2 on microglial survival and a stimulatory effect on the production of inflammatory cytokines, which is not dependent on the presence of TREM2 or DAP12 (Zhong et al., 2017). Now the authors extend their investigation to the effects of administering sTREM2 (in different ways) and show a positive effect on microglia recruitment to the sites of amyloid plaques, leading to a reduction in overall plaque size and pointing to a protective role of wild-type sTREM2.
It would have been interesting to see, in the manuscript, if mutant forms of sTREM2 (e.g., p.R47H) would have shown a similar loss-of-function phenotype as has been described for the full-length mutant receptor. So far the full-length receptor has been associated with beneficial functions (e.g., microglial recruitment to the amyloid plaque sites) and a proof-of-principle experiment indicated that stabilizing full-length TREM2 results in increased phagocytic activity (Kleinberger et al., 2014). Furthermore, increased production of sTREM2 due to the presence of the p.H157Y variant has been associated with increased risk for developing AD in a Han Chinese population (Jiang et al., 2016). Studies like this one by Zhong and colleagues are crucial to understand the functions of both full-length receptor and its cleaved ectodomain and are extremely instrumental in helping to design strategies to therapeutically target TREM2.
References:
Schlepckow K, Kleinberger G, Fukumori A, Feederle R, Lichtenthaler SF, Steiner H, Haass C. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med. 2017 Oct;9(10):1356-1365. PubMed.
Thornton P, Sevalle J, Deery MJ, Fraser G, Zhou Y, Ståhl S, Franssen EH, Dodd RB, Qamar S, Gomez Perez-Nievas B, Nicol LS, Eketjäll S, Revell J, Jones C, Billinton A, St George-Hyslop PH, Chessell I, Crowther DC. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer's disease-associated H157Y variant. EMBO Mol Med. 2017 Oct;9(10):1366-1378. PubMed.
Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, Norton JB, Hsu S, Harari O, Cai Y, Bertelsen S, Goate AM, Cruchaga C. Coding variants in TREM2 increase risk for Alzheimer's disease. Hum Mol Genet. 2014 Nov 1;23(21):5838-46. Epub 2014 Jun 4 PubMed.
Brendel M, Kleinberger G, Probst F, Jaworska A, Overhoff F, Blume T, Albert NL, Carlsen J, Lindner S, Gildehaus FJ, Ozmen L, Suárez-Calvet M, Bartenstein P, Baumann K, Ewers M, Herms J, Haass C, Rominger A. Increase of TREM2 during Aging of an Alzheimer's Disease Mouse Model Is Paralleled by Microglial Activation and Amyloidosis. Front Aging Neurosci. 2017;9:8. Epub 2017 Jan 31 PubMed.
Suárez-Calvet M, Araque Caballero MÁ, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C, Dominantly Inherited Alzheimer Network. Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016 Dec 14;8(369):369ra178. PubMed.
Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, Huang R, Wang D, Li X, Wu L, Jia L, Zheng H, Painter M, Atagi Y, Liu CC, Zhang YW, Fryer JD, Xu H, Bu G. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017 Mar 6;214(3):597-607. Epub 2017 Feb 16 PubMed.
Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
Jiang T, Tan L, Chen Q, Tan MS, Zhou JS, Zhu XC, Lu H, Wang HF, Zhang YD, Yu JT. A rare coding variant in TREM2 increases risk for Alzheimer's disease in Han Chinese. Neurobiol Aging. 2016 Jun;42:217.e1-3. Epub 2016 Mar 3 PubMed.
Washington University School of Medicine
The University of Hong Kong
The paper is remarkable as it implies that even a single dose of sTREM2 protein can ameliorate AD pathology by microglia. It would be important to study the effects of repeated injection of sTREM2 at different phases of AD pathology to test the potential use of sTREM2 as a therapeutic agent. In addition, it would also be interesting to see which cell types in the CNS can be affected by administration of soluble TREM2, since sTREM2 possesses the tendency to contact any cell type in the brain, as reported by the authors. An exciting goal for future studies is the identification of the receptors of sTREM2 in the CNS.
Indiana University School of Medicine
Indiana University
Zhong and colleagues have recently reported a remarkable series of findings that demonstrate that soluble forms of TREM2 exhibit cell-autonomous actions and act to mitigate AD pathogenesis in mouse models of AD. These observations serve to focus attention on the biological roles of soluble TREM2 (sTREM2). There is compelling evidence demonstrating that CSF levels of sTREM2 correlate with AD progression, and there are genetic links between the abundance of these species and risk of AD. The discovery that soluble forms of the receptor are generated through proteolytic cleavage of its extracellular domain, and that sTREM2 is found at increased levels in the CSF in AD patients, provides a potential biomarker for AD pathogenesis. However, the biological significance of sTREM2 in CSF is not understood and is poorly explored. The possibility that sTREM2 has cell autonomous actions is provocative and potentially of substantial importance. The present study is of particular interest, but ultimately unsatisfying in that their findings are largely phenomenological and the paper does not engage many of the central issues raised by their studies.
It has been speculated that the proteolytic cleavage of Trem2, liberating its extracellular domain into the interstitial fluid, represents the functional inactivation of the full-length, membrane-associated receptor, which is thought to be deleterious with respect to disease pathogenesis. Moreover, sTREM2 species, acting as a decoy, might logically suppress microglial TREM2 activation by binding its ligands. Indeed, therapeutics directed at inhibiting this cleavage are under development. However, the present paper makes the opposite argument, whereby sTREM2 elicits a broad range of salutary actions by microglia that mimic those associated with activation of the full-length, membrane-associated TREM2.
This present study by Zhong and colleagues follows their previous report that sTREM2 species exhibit microglial-directed, cell-autonomous actions, stimulating pro-inflammatory gene expression and enhancing microglial survival and proliferation (Zhong et al., 2017). The authors now report a large number of findings which result in a hypothesis that is hard to fathom.
They show that injection of a purified form of sTREM2 stimulates a variety of microglial phenotypic changes and a reduction in amyloid plaque load and dystrophic neurites. These effects are analogous to that observed with direct microglial TREM2 activation. They previously have shown that sTREM2 actions on microglia do not involve the membrane-bound, full-length receptor and elicited a very robust pro-inflammatory response. This latter response is, again, hard to reconcile with their hypothesis. It is unknown and unexplored how sTREM2 might interact with microglia in such an autocrine loop.
It is difficult to deduce from the paper that the sTREM2 injected (6ug) comprised the entire extracellular domain (1-171) and resulted in supraphysiological concentrations of sTREM2. These experiments included only a vehicle control, and not an inactive protein control. It is only in a subset of subsequent studies that appropriate controls are performed with the biologically relevant sTREM2 (1-157) species and a heat-inactivated control. A different range of concerns arise from studies in which sTREM2 is overexpressed in the 5XFAD mice using an AAV construct that includes the entire extracellular domain, EGFP, and a tag that is expressed at levels about 500-fold over endogenous levels. In this setting sTREM2 protein is expressed largely from neurons, a cell type that does not express TREM2. I don’t think these experiments are interpretable.
Colonna and colleagues reported sTREM2 was found to be associated with neurons and plaques, a finding reproduced in the present study (Song et al., 2018). These data suggest that there is robust expression of neuronal TREM2 ligands to which sTREM2 binds. It is curious that this observation was not vigorously investigated beyond determination that in vitro neuronal electrophysiological characteristics are unaffected by sTREM2. Zhong et al. find a remarkable effect of sTREM2 on LTP and argue that this is a microglial-mediated response, although it is not clear how this might be achieved.
In summary, although this study reports very compelling datasets, the excitement surrounding these findings is undercut by the unusual experimental paradigms and confounding experimental design.
Overall, the suggestion that sTREM2 has cell-autonomous actions is both provocative and important. Indeed, the broad range of potential actions reported by Zhong et al. puts the question of the biology of TREM2 in an entirely new perspective. However, the lack of a cogent and compelling hypothesis supported by the data undermines enthusiasm for this paper.
References:
Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, Huang R, Wang D, Li X, Wu L, Jia L, Zheng H, Painter M, Atagi Y, Liu CC, Zhang YW, Fryer JD, Xu H, Bu G. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017 Mar 6;214(3):597-607. Epub 2017 Feb 16 PubMed.
Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med. 2018 Mar 5;215(3):745-760. Epub 2018 Jan 10 PubMed.
Make a Comment
To make a comment you must login or register.