Too Clingy: Extra Hydrogen Bond Prompts Protein Aggregation
Quick Links
Protein low-complexity domains (LCDs) stack together to form dynamic liquid droplets within the cell; alas, some pathogenic mutations in these regions encourage aggregation beyond this temporary physiological state. What flips the switch? Something as seemingly ephemeral as one little hydrogen bond, according to researchers led by Steven McKnight and Glen Liszczak, University of Texas Southwestern Medical Center, Dallas. In the July 1 Science, they reported that mutations in proline—which free the backbone nitrogen-to-hydrogen bond—unduly stabilize LCD peptide aggregates. Experimentally capping this nitrogen normalized peptide solubility. This was true for point mutations in tau, neurofilament light (NfL), and the RNA-binding proteins TDP-43 and hnRNPA2. All are tied to neurodegenerative diseases.
- Proline mutations enable a nitrogen-to-hydrogen bond in the protein's backbone.
- This strengthens cross-β sheets between peptides, forming fibrils.
- Methylating the main-chain nitrogen normalizes protein structure.
“This work [provides] a structural connection of gene mutation to pathology,” wrote David Eisenberg, University of California, Los Angeles (full comment below).
“These in vitro studies suggest that … peptide backbone hydrogen bonds can influence aberrant phase transitions of LCDs and could potentially be tuned to control disease-linked protein self-association and aggregation,” wrote Dorothee Dormann, Johannes Gutenberg-Universität Mainz, Germany (full comment below). Ben Wolozin at Boston University agreed, noting that “this is one of the cumulative factors that lead to TDP-43 aggregation.”
LCDs of TDP-43, tau, and hnRNA2 self-aggregate by forming labile cross-β-sheets, enabling the proteins to partially slip out of the cytosol as liquid droplets, a bit like oil suspended in water (Mar 2019 news; Aug 2017 news; Feb 2018 news). What drives this liquid-liquid phase separation?
First author Xiaoming Zhou suspected backbone hydrogen bonding because it is crucial for β-sheet formation. He created 23 variants of TDP-43’s LCD region, a 150-amino-acid peptide, each with one changed amino acid in the conserved portion of the protein. The synthetic amino acids contained a methylated backbone nitrogen to prevent intermolecular hydrogen bonding. Zhou then used microscopy to analyze each peptide’s ability to undergo phase separation in a neutral isotonic buffer.
Nine variants failed to coalesce into liquid droplets. All the mutated residues fell within the region that forms the β-sheet core (Dec 2021 news). “That removal of a single backbone hydrogen bond could disrupt the entire structure shows just how weak—and therefore dynamic and reversible—the self-association is,” Gregory Petsko, Harvard Medical School, Boston, and Scott Small, Columbia University, New York, wrote in an editorial in the same issue of Science. The researchers believe that hydrogen bonds from the peptide’s backbone are crucial for β-sheet self-aggregation and phase separation.
Amino acid side chains may also influence peptide structure. Zhou systematically swapped each amino acid in TDP-43 LCD’s conserved region to glycine. Intriguingly, the most profound change occurred in the proline 320 mutant, which precipitated into tangles rather than coalescing into liquid droplets. This mutation freed the residue’s main-chain nitrogen-to-hydrogen bond. When the scientists capped this nitrogen by incorporating an N-methyl glycine into the LCD peptide, droplets formed again. The authors concluded that freeing this backbone nitrogen stabilized labile aggregates so they precipitated (see image below).
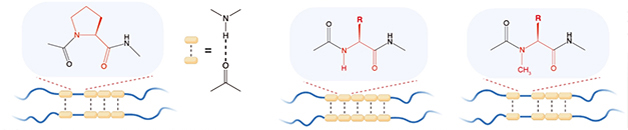
Hydrogen Velcro? Backbone hydrogen bonding (dotted lines) lightly holds together low-complexity domains (blue squiggles). These domains remain labile due to the occasional proline (left), which cannot H-bond. Substituting any other residue allows main-chain H-bonding (middle) to stabilize the cross-β-sheet. This is reversed by methylating the backbone nitrogen (right) to restore H-bonding. [Courtesy of Zhou et al., Science, 2022.]
Could mutations in the DNA sequence encoding proline explain why variants in other aggregate-prone proteins increase the risk of neurodegenerative disease? Indeed, converting proline to another amino acid within LCDs of tau is known to cause tauopathies, proline swaps in NfL cause the hereditary peripheral neuropathy Charcot-Marie-Tooth disease, and, in hnRNPA2, amyotrophic lateral sclerosis and the bone disorder Paget’s disease (Jordanova et al., 2003; Shin et al., 2008; Qi et al., 2017).
Specifically, peptides of tau's mid-domain that harbor either P301S, P301T, or P301L had a strong propensity to aggregate and sequester thioflavin-T dye, which shifts its fluorescence after clinging to aggregated proteins. Likewise, six NfL proline mutants readily polymerized to form amorphous tangles rather than wild-type filaments. The LCD fragment of hnRNPA2 containing P298L aggregated and melded into misshapen liquid droplets, agreeing with previous research (Feb 2018 news). For each of these mutant proteins, incorporating N-methylated amino acids to block the extra hydrogen bond reversed its stability and solubility to that of the wild-type (see image below).

Out of Phase. Pathogenic proline mutations trigger NfL to tangle haphazardly (top left), tau to aggregate when it shouldn't (middle left), and hnRNPA2 to coalesce into liquid droplets that look misshapen (bottom left). Methylating the main-chain nitrogen of the mutated amino acid restored all three proteins to their wild-type conformations (right panels). [Courtesy of Zhou et al., Science, 2022.]
One caveat of these experiments? They involved only the LCD fragment of any given protein, not the complete sequence. “What is very clear with a small peptide like this is more ambiguous when studying the entire protein,” Wolozin told Alzforum. How pathologically relevant these findings are remains unclear.
Dan Li, Shanghai Jiao Tong University, China, wondered about detecting the labile cross-β-sheets in cells or tissue. While not providing a direct intracellular view, deep mutation scanning enables systematic study of thousands of variants on protein structure and aggregation within cells (Starita et al., 2017). “It is possible to reveal structural patterns and aggregation state in vivo of full-length, LCD-containing proteins and determine their effect on cellular fitness,” agreed Benedetta Bolognesi, Institute for Bioengineering of Catalonia, Barcelona, Spain (full comments below).
Using deep mutation scanning, Bolognesi found that, of 52,000 mutations within the LCD of TDP-43, the conserved region was a hotspot for creating toxic mutants that formed liquid-like droplets rather than solid aggregates (Bolognesi et al., 2019). Intriguingly, the pattern of interactions between LCD mutations suggested two structures in this region: a β-sheet at residues 311 to 315 and an α-helix at 324 to 331.
Researchers are debating which secondary structure TDP-43’s conserved region contorts into. Some have reported that TDP-43’s α-helices, not β-sheets, drive liquid-liquid phase separation (Sep 2016 news). This raises the question of whether cross-β-sheets truly control LCD phase separation, as Zhou’s study did not include structural experiments. “An alternative possibility could be that the introduced backbone methyl groups interfere with the helical conformation and thereby interfere with phase separation of TDP-43,” Dormann wrote. The labs directly involved in this debate declined to comment.—Chelsea Weidman Burke
References
News Citations
- More Evidence for Distinct TDP-43 Droplets
- More Droplets of Tau
- Are Reversible Amyloids Behind Liquid-Liquid Phase Separation?
- Double Spiral Sets TDP-43 Apart from Other Amyloids
- How Does a Neuron Avoid Aggregation of Liquid Protein Droplets?
- Helical Tail Holds Sway Over TDP-43 Packaging
Mutations Citations
Paper Citations
- Jordanova A, De Jonghe P, Boerkoel CF, Takashima H, De Vriendt E, Ceuterick C, Martin JJ, Butler IJ, Mancias P, Papasozomenos SC, Terespolsky D, Potocki L, Brown CW, Shy M, Rita DA, Tournev I, Kremensky I, Lupski JR, Timmerman V. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain. 2003 Mar;126(Pt 3):590-7. PubMed.
- Shin JS, Chung KW, Cho SY, Yun J, Hwang SJ, Kang SH, Cho EM, Kim SM, Choi BO. NEFL Pro22Arg mutation in Charcot-Marie-Tooth disease type 1. J Hum Genet. 2008;53(10):936-940. Epub 2008 Aug 29 PubMed.
- Qi X, Pang Q, Wang J, Zhao Z, Wang O, Xu L, Mao J, Jiang Y, Li M, Xing X, Yu W, Asan, Xia W. Familial Early-Onset Paget's Disease of Bone Associated with a Novel hnRNPA2B1 Mutation. Calcif Tissue Int. 2017 Aug;101(2):159-169. Epub 2017 Apr 7 PubMed.
- Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, Shendure J, Fowler DM. Variant Interpretation: Functional Assays to the Rescue. Am J Hum Genet. 2017 Sep 7;101(3):315-325. PubMed.
- Bolognesi B, Faure AJ, Seuma M, Schmiedel JM, Tartaglia GG, Lehner B. The mutational landscape of a prion-like domain. Nat Commun. 2019 Sep 13;10(1):4162. PubMed.
Further Reading
Papers
- Zhou X, Lin Y, Kato M, Mori E, Liszczak G, Sutherland L, Sysoev VO, Murray DT, Tycko R, McKnight SL. Transiently structured head domains control intermediate filament assembly. Proc Natl Acad Sci U S A. 2021 Feb 23;118(8) PubMed.
News
- Death by Goo: TDP-43 Gels Paralyze Proteasomes in Neurons
- Stress Granules: No Incubator for Inclusions, After All?
- Liquid Phase Transition: A Deluge of Data Points to Multiple Regulators
- Structural Biology Sheds Light on Regulation of Liquid-Liquid Phase Transition
- Paper Alert: Stress Granules Get Tangled Up in Tau
- Protein Liquid-Liquid Phase Transitions: The Science Is About to Gel
- Islands of Tau Coat and Protect Cytoskeleton
- Disease Mutations Zip Lock Stress Granules in Proteinopathy, ALS
Primary Papers
- Zhou X, Sumrow L, Tashiro K, Sutherland L, Liu D, Qin T, Kato M, Liszczak G, McKnight SL. Mutations linked to neurological disease enhance self-association of low-complexity protein sequences. Science. 2022 Jul;377(6601):eabn5582. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UCLA
Geneticists have found mutations in genes that encode proteins associated with several neurodegenerative conditions, including Alzheimer’s, Parkinson’s, frontotemporal degeneration, and ALS. But do these mutated proteins actually cause disease, and if so, how? Confidently distinguishing cause and effect from mere association requires identifying a mechanism of action. The carefully planned and executed biochemical studies reported by Zhou et al. support mechanisms by which these proteins form toxic aggregates leading to disease. Of particular importance, these mechanism operate through specific three-dimensional structures, parallel, in-register β-sheets, of the type first described by Linus Pauling and Robert Corey 70 years ago.
Such β-sheets have been known for some years to be associated with neurodegeneration. What is new here are similar, but less stable forms of β-sheets that are formed between domains of proteins formed from a restricted repertoire of amino acid residues. Such domains are termed low-complexity domains. Protein scientists have been aware of low-complexity domains for years, but the McKnight lab has led the way to understanding the structural basis for the interaction of proteins by forming weak β-sheets via their low-complexity domains. Their work explains the mechanism—a structural connection of gene mutation to pathology--illuminating the origins of neurodegeneration.
JGU Mainz
This study proposes that individual hydrogen bonds between polypeptide backbones of low-complexity domains (LCDs) contribute to self-association and aberrant phase transitions of neurodegenerative disease-linked proteins, such as TDP-43, Tau, and hnRNP-A2. The authors used an interesting native chemical ligation strategy, chemically linking together different modified peptides to assemble disease-linked LCDs. Using peptides with single methyl-capped backbone nitrogen atoms at different positions, they analyzed the impact of eliminating individual hydrogen bonds from different positions within the TDP-43 LCD. They found that removing hydrogen bonds from positions 321–329 in the TDP-43 LCD strongly reduces the capacity of the TDP-43 LCD to phase separate, as measured by turbidity assays. The authors attribute this reduction to the disruption of cross-β interactions in this region, although structural studies were not performed in the current study.
An alternative model posits that this region adopts an α-helical conformation (Conicella et al., 2016; Conicella et al., 2020), hence an alternative possibility could be that the introduced backbone methyl groups interfere with this helical conformation and thereby interfere with phase separation of TDP-43. These alternative possibilities remain to be addressed by future structural studies.
Zhou et al. furthermore address the contribution of different amino acid side chains within the TDP-43 LCD to phase separation and aggregation. Using glycine-scanning mutagenesis, they individually replaced the amino acids in the conserved 315–340 region of the TDP-43 LCD with glycine and analyzed condensate morphology by microscopy. This revealed the importance of a particular proline residue, P320, as an “interaction breaker,” as mutant TDP-43 LCD with a P320G mutation massively aggregated instead of forming round condensates.
Additional amino acids in the 324–330 region (AAAQAAL) seemed to have a similar interaction-reducing and aggregation-buffering effect, whereas other amino acids, e.g., W334 and methionines M336, M337, M339, appeared to have the oppositive effect and likely enhance TDP-43 LCD self-association, as their mutation to glycine led to larger, rounder TDP-43 condensates, indicative of a more liquid-like, dynamic behavior. This hypothesis remains speculative, as the material state and dynamicity of the formed condensates was not experimentally addressed in the current study. An interesting side-finding that is, however, not mechanistically followed up in the current study, is that the P320G mutation leads to cytoplasmic aggregation of TDP-43 in cells. The effect of the introduced mutations on putative cross-β versus α-helical structures also remains to be addressed.
Finally, based on their TDP-43 findings, the authors scrutinized other disease-associated proline mutations in disease-linked proteins with LCDs, e.g., in neurofilament light chain (NfL), Tau and hnRNP-A2. They found that certain disease-linked proline mutations in these LCDs have a similar aggregation-promoting effect and that introducing methyl groups into the peptide backbone nitrogens at these positions can abrogate or reduce LCD aggregation, as measured by Thioflavin T incorporation.
Taken together, the in vitro studies performed by Zhou and colleagues suggest that, in addition to specific amino acid side chains or post-translational modifications thereon, peptide backbone hydrogen bonds can influence aberrant phase transitions of LCDs and could potentially be tuned to control disease-linked protein self-association and aggregation.
References:
Conicella AE, Zerze GH, Mittal J, Fawzi NL. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure. 2016 Sep 6;24(9):1537-49. Epub 2016 Aug 18 PubMed.
Conicella AE, Dignon GL, Zerze GH, Schmidt HB, D'Ordine AM, Kim YC, Rohatgi R, Ayala YM, Mittal J, Fawzi NL. TDP-43 α-helical structure tunes liquid-liquid phase separation and function. Proc Natl Acad Sci U S A. 2020 Mar 17;117(11):5883-5894. Epub 2020 Mar 4 PubMed.
Shanghai Jiao Tong University
Phase separation of different proteins has been widely observed in a variety of biological processes. Among them, several amyloid proteins including TDP-43, FUS, and hnRNPA1 were found to form dynamic liquid-like droplets, and further condense into irreversible amyloid fibrils, which are closely associated with different neurodegenerative diseases including ALS and FTD. Numerous studies reveal that the irreversible amyloid fibrils formed by these pathological proteins exhibit typic cross-β structures.
Despite that, several groups have reported that segments of TDP-43, FUS and hnRNPA1 can form labile cross-β structures that are much less stable than the typical steric-zipper cross-β structures found in irreversible fibrils (Hughes et al., 2018; Murray et al., 2017; Luo et al., 2018), it remains debated in the field whether the labile cross-β interaction plays a role in the dynamic liquid phase formed by these proteins.
In this study, McKnight and co-workers tackle this important question by using the backbone nitrogen methylated TDP-43 LCD, which can block the main-chain intermolecular hydrogen bonding essential for cross-β structure with minimal effect on protein side-chains. By using a peptide ligation and semisynthesis method, the authors were able to obtain more than 20 different semisynthetic variants of the TDP-43 LCD with either single or multiple methylated backbone nitrogen atoms.
They found that, within residues 321-329 of TDP-43 LCD previously identified to form the cross-β structure, methyl capping of each single backbone nitrogen can severely impair the phase separation capability of TDP-43 LCD. This supports the notion that cross-β interaction mediated by this region is important for mediating TDP-43 LCD phase separation.
Moreover, the authors found that the fine balance of the cross-β interaction is important for the dynamic protein phase separation. Disease-associated mutations that strengthen the cross-β interaction can promote formation of irreversible amyloid fibrils. In line with this finding, the Eisenberg group recently reported that disease mutations on intermediate filament protein keratin-8 (KRT8) can strengthen the otherwise labile cross-β interaction and promote pathological aggregation of KRT8 involved in alcoholic steatohepatitis (Murray et al., 2022).
I think the following questions may need to be addressed in the future: (1) How general is this phenomenon in different proteins involved in phase separation and aggregation? From the computational screening for pathological mutations done by the Eisenberg group (Murray et al., 2022), very limited proteins were identified. This could be caused by the bias of the structural template used for computational searching. (2) Can we directly detect the labile cross-β structures in cell models or brain tissues by developing antibody or specific dye molecules?
References:
Hughes MP, Sawaya MR, Boyer DR, Goldschmidt L, Rodriguez JA, Cascio D, Chong L, Gonen T, Eisenberg DS. Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science. 2018 Feb 9;359(6376):698-701. PubMed.
Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, Tycko R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell. 2017 Sep 16; PubMed.
Luo F, Gui X, Zhou H, Gu J, Li Y, Liu X, Zhao M, Li D, Li X, Liu C. Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat Struct Mol Biol. 2018 Apr;25(4):341-346. Epub 2018 Apr 2 PubMed.
Murray KA, Hughes MP, Hu CJ, Sawaya MR, Salwinski L, Pan H, French SW, Seidler PM, Eisenberg DS. Identifying amyloid-related diseases by mapping mutations in low-complexity protein domains to pathologies. Nat Struct Mol Biol. 2022 Jun;29(6):529-536. Epub 2022 May 30 PubMed.
IBEC
This is a clever approach that allows one to have a neat readout of the phenotypic outcome of main chain hydrogen bonding. I enjoyed reading this paper.
The authors imply that, at least for this small set of proteins, the same interactions driving LLPS are those that then become stronger upon mutation and drive aggregation into fibrils. I.e. that, albeit more labile, the cross-β structure exists already within (functional?) droplets. I find this extremely interesting, and think it will generate a good amount of discussion in the LLPS field, which is healthy.
If this is the case, then evolution must have put in place strong regulatory mechanisms to control this process and avoid aggregation. This would include specific sequence patterns in the areas surrounding these stretches, as well as cellular machinery able to counteract the aggregation process, at the expense of a lot of energy. Is there evidence of these patterns or mechanisms?
On the other hand, this conclusion may also be partially biased by the fact that a large portion of the flanking regions are missing from these peptides, which therefore lack the opportunity to establish other kinds of interactions.
I am particularly intrigued by the results on TDP-43, as just three years ago my lab, together with Ben Lehner’s lab, identified this exact conserved region as a hotspot of mutational effect when measuring the toxicity of TDP-43 sequences by deep mutational scanning (Bolognesi et al., 2019). Seeing the prominent role of Pro20 in maintaining the labile connotation of these structures reminds me that this residue is the only position in our dataset where all mutations have a similar effect (relieving toxicity in yeast), and where mutations are likely to promote formation of aggregates likely to be solid-like rather than liquid-like, in line with what Zhou et al. suggest here.
The observations on prolines and proline mutations across different systems are exciting and raise important questions regarding their evolutionary implications. Is it true that Pro residues are conserved in, or in proximity across, LCDs? The authors show some cool examples in NTF, and a systematic evolutionary analysis across all known LCDs would be helpful. Are mutations in these prolines recurrent in disease beyond the cases shown? How frequent are they?
Prolines can’t engage in main chain H-bond, but one can also simply say they impose strong overall constraints as they are incompatible with any type of secondary structure. This means they would impact cross-β quaternary arrangements, but also all other types of local structure in this region. Finally, the type of amino acid introduced upon mutation should also contribute. One could argue that a valine could strengthen a cross-β conformation, while burying an aspartic in that interface would be borderline impossible.
Not a crucial issue, but one limitation I see is the following. Upon N-capping and denial of specific hydrogen bonds, the range of resulting phenotypes is actually quite diverse across the four proteins studied: in vitro mis-shaped droplets, amorphous aggregates, amyloid fibrils, intracellular aggregation. It is hard for me to be sure that all of the above are part of one unique common pathway.
MAVEs (Multiplexed Assays of Variant Effect), also called deep mutational scanning (Starita et al., 2017), are allowing us to test many of these hypotheses in a comprehensive and systematic way rather than in a hypothesis-driven way. Provided a read-out exists [e.g., cell toxicity (Bolognesi et al., 2019), amyloid nucleation (Seuma et al., 2021), protein stability (Faure et al., 2022; Matreyek et al., 2018)], by measuring the impact of thousands of mutations on a specific biological mechanism in parallel, it will be possible to reveal those properties or structural patterns that in vivo and in full-length proteins determine the aggregation state of LCD-containing proteins and their effect on cellular fitness.
References:
Bolognesi B, Faure AJ, Seuma M, Schmiedel JM, Tartaglia GG, Lehner B. The mutational landscape of a prion-like domain. Nat Commun. 2019 Sep 13;10(1):4162. PubMed.
Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, Shendure J, Fowler DM. Variant Interpretation: Functional Assays to the Rescue. Am J Hum Genet. 2017 Sep 7;101(3):315-325. PubMed.
Seuma M, Faure AJ, Badia M, Lehner B, Bolognesi B. The genetic landscape for amyloid beta fibril nucleation accurately discriminates familial Alzheimer's disease mutations. Elife. 2021 Feb 1;10 PubMed.
Faure AJ, Domingo J, Schmiedel JM, Hidalgo-Carcedo C, Diss G, Lehner B. Mapping the energetic and allosteric landscapes of protein binding domains. Nature. 2022 Apr;604(7904):175-183. Epub 2022 Apr 6 PubMed.
Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, Kircher M, Khechaduri A, Dines JN, Hause RJ, Bhatia S, Evans WE, Relling MV, Yang W, Shendure J, Fowler DM. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet. 2018 Jun;50(6):874-882. Epub 2018 May 21 PubMed.
Make a Comment
To make a comment you must login or register.