Can ‘Cryptic Peptides’ Peg People with TDP-43 Pathology?
Quick Links
Could a biomarker for TDP-43 pathology be on the horizon? This RNA/DNA-binding protein accumulates in nearly all people with amyotrophic lateral sclerosis, and almost half of those with frontotemporal dementia, but can only be detected at autopsy. Two new bioRxiv preprints may change that.
In one, uploaded January 23, Michael Ward at the National Institute of Neurological Disorders and Stroke, Bethesda, Maryland, Pietro Fratta at University College London, Leonard Petrucelli at the Mayo Clinic in Jacksonville, Florida, and colleagues describe the consequences of RNA mis-splicing errors caused by loss of TDP-43 from the nucleus, as occurs in TDP-43 proteopathies. These errors result in transcripts that contain extra pieces, called “cryptic exons.” They are usually degraded. However, the researchers found that a small number of such transcripts are made into proteins. They detected eight such “cryptic peptide”-containing proteins in the cerebrospinal fluid of people with ALS/FTD, suggesting they could be biomarkers of the disease.
- In TDP-43 proteopathy, some mis-spliced transcripts are translated into protein.
- These proteins are detected in the CSF of patients but not controls.
- They may flag presymptomatic disease and predict onset.
“This is a remarkable feat of science by a fantastic group of experts,” Peter Nelson at the University of Kentucky, Lexington, told Alzforum. Sami Barmada at the University of Michigan, Ann Arbor, wrote, “This is one of the clearest demonstrations that changes at the RNA level in models of disease have substantial and consequential impacts at the protein level.”
The second manuscript, uploaded January 24, delved deeper into the biomarker potential. Scientists led by Philip Wong at Johns Hopkins University School of Medicine, Baltimore, report that they detected a cryptic peptide version of histone-binding protein HDGFL2 in the CSF of people with a C9ORF72 expansion, even before they developed symptoms of ALS or FTD. These expansions cause TDP-43 to aggregate in the cytosol, sucking it out of the nucleus (Dejesus-Hernandez et al., 2011; Ryan et al., 2022).
“These converging studies establish long-sought prognostic TDP-43-dependent protein biomarkers, which will transform not only the field of ALS-FTD, but other human disease exhibiting TDP-43 proteinopathy,” Wong predicted.

From Cryptic Exon to Peptide. In the absence of nuclear TDP-43, splicing errors lead to new sequences (red) in some transcripts. Some of these transcripts are made into proteins with new domains. [Courtesy of Seddighi et al., 2023.]
Wong and colleagues first reported TDP-43’s role in keeping cryptic exons out of transcripts (Aug 2015 news). TDP-43 affects splicing of thousands of genes, and as it is lost from the nucleus, such errors accumulate. Typically, cryptic exons disrupt the normal reading frame of a transcript, or add a premature stop codon. Cells recognize these faulty transcripts and dispose of them before they can be translated. Subsequent studies by several groups identified specific examples of this, finding that cryptic exons in transcripts for the microtubule-binding protein stathmin-2, and the axonal regulator UNC13A, squelched expression of those proteins (Dec 2018 conference news; Jan 2019 news; Apr 2021 news).
Are transcripts with cryptic exons always degraded? To investigate, joint first authors Sahba Seddighi and Yue Qi in Ward’s lab, and Anna-Leigh Brown in Fratta’s, generated glutamatergic neurons from human iPSCs that had their TDP-43 gene knocked out by CRISPR. As expected, RNA splicing ran amok in these neurons. The authors identified 233 abnormally spliced transcripts, most of which contained cryptic exons, though a few skipped exons instead, or included entire introns. Most of these bungled transcripts were degraded, as shown by the low levels of their protein product compared to neurons with normal TDP-43 expression.

Wheels of Peptides. Glutamatergic neurons lacking TDP-43 made new versions of 12 proteins (names colored). As depicted in the outer wheel, some had peptides added in-frame (blue-gray outer circle), some with out-of-frame peptides (green), others with whole introns retained (mud gray), or with exons skipped entirely (purple). The inner wheel represents the number of individual trypsin-digested peptides found for each gene. These are colored to match the gene name. [Courtesy of Seddighi et al., 2023.]
Some were translated, however. Mass spectrometry analysis of protein from these neurons pulled out 12 that expressed cryptic peptides: HDGFL2, MYO18A, CAMK2B, DNM1, ACTL6B, KCNQ2, MYO1C, ADGRB1, AGRN, SYT7, KALRN, RSF1 (image at right). This list is not comprehensive, as different experiments identified other cryptic peptides. In most of the 12, though, the cryptic exon or abnormal splice site maintained the transcript reading frame, creating a nearly normal version of the protein with an added domain. Such in-frame cryptic exons tend to escape cell quality-control mechanisms, and thus are more likely to be translated. However, for SYT7, KALRN, and RSF1, the cryptic exon altered the reading frame and introduced premature stop codons, producing proteins truncated after the new domain.
These new domains may affect protein function. The authors immunoprecipitated HDGFL2 from TDP-43 knockout neurons, and found it brought down different proteins than did HDGFL2 from control neurons. Out of 178 protein partners, 16 were enriched and 13 depleted in TDP-43 knockout neurons. The former tended to regulate mRNA splicing and DNA repair, perhaps to compensate for loss of TDP-43, while the latter were mostly cytoskeleton regulators. This suggests cryptic peptides could change the biology of the cell, potentially shedding light on previously unknown disease mechanisms, Ward noted.
Fen-Biao Gao at the University of Massachusetts Medical School thinks the data support the idea that correcting multiple transcripts affected by TDP-43 may be necessary to ameliorate disease (Halim and Gao, 2022). “A major challenge for the future is to identity the subset of TDP-43 targets, if they exist, that may be responsible for the bulk of the consequences of loss of TDP-43 function,” Gao wrote to Alzforum (full comment below).
Do cryptic peptides occur in people? Using mass spec, the scientists measured four dozen proteins, known to be abundant in ALS CSF, and that could contain cryptic peptides. In 12 or more of 15 samples from ALS patients, eight of these proteins—HDGFL2, MYO18A, RSF1, SYT7, SYNE1, IGLON5, SYN3, TRRAP—contained cryptic peptides, suggesting they could be biomarkers of the disease. The list varies from that of de novo peptides found in cultured neurons because not all proteins make it into CSF in large enough quantity to be detected, while other proteins are more abundant there than in neurons, Ward said. He noted that investigations of other cell types, such as inhibitory neurons or astrocytes, might turn up additional cryptic peptides that could be better disease markers.
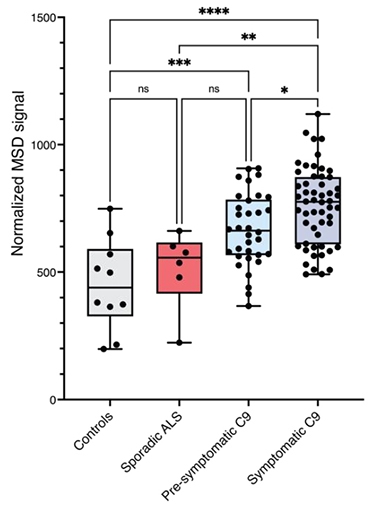
A Biomarker for ALS? As determined by a meso-scale discovery (MSD) immunoassay, cryptic peptide-containing HDGFL2 was higher in CSF of familial and sporadic ALS patients than in control CSF, even at presymptomatic stages. [Courtesy of Irwin et al., 2023.]
Separately, Wong and colleagues focused on whether cryptic peptides could be biomarkers of disease. Rather than a large proteomic analysis, as Seddighi and colleagues had done, first author Katherine Irwin generated monoclonal antibodies to cryptic peptides in just five proteins: HDGFL2, ACTL6B, AGRN, EPB41L4A, and SLC24A3. She selected these because the normal proteins are highly expressed in brain, and their cryptic exons were predicted to be in-frame and produce exposed epitopes that might be easily detected by antibodies. The antibody against HDGFL2 was particularly promising. It detected a cryptic peptide-containing version of the protein in HeLa cells lacking TDP-43.
The authors developed a sandwich ELISA and used it to quantify the amount of cryptic HDGFL2 in CSF samples from 31 C9ORF72 carriers with symptoms of ALS or FTD, and from 15 asymptomatic carriers, who were enrolled in a natural history study at the NIH (Offit et al., 2020). Participants donated two samples, on average, over 2.5 years. The amount of cryptic HDGFL2 was significantly higher in carriers, even those who were presymptomatic, than in CSF samples from 10 controls. Wong and colleagues had earlier found that cryptic exons show up in transcripts in postmortem Alzheimer’s disease brain as soon as TDP-43 abandons the nucleus, and before it forms visible deposits, suggesting those RNAs could be early markers of pathology (Sun et al., 2017).
“Since the delay between first symptoms and a diagnosis of ALS can be frustratingly long, the potential to find a biomarker that works presymptomatically would be very valuable,” noted Steven Finkbeiner at the Gladstone Institutes in San Francisco (comment below).
However, the amount of cryptic HDGFL2 did not correlate with symptom severity, nor with deterioration over the 2.5 years of the study. Moreover, although levels were slightly higher in symptomatic than presymptomatic carriers, among the symptomatic group, they tended to be lower the longer that person had had symptoms. This suggests expression peaks early in the symptomatic stage, and then falls. This is the opposite pattern to the neurodegeneration marker NfL in ALS/FTD, which climbs steadily as symptoms worsen (image below). Might this difference help clinicians better identify disease stages?

Diverging Trajectories. The opposite longitudinal trajectories of cryptic HDGFL2 and NfL could help predict when symptoms will appear, with ratios around one indicating onset is imminent. [Courtesy of Irwin et al., 2023.]
Indeed, the ratio of NfL to cryptic HDGFL2 distinguished presymptomatic and symptomatic carriers in this cohort. Overall, 82 percent of presymptomatic people had a ratio below one, while 97 percent of symptomatic people were above one. Notably, the presymptomatic volunteer with the highest NfL/cryptic HDGFL2 ratio developed movement problems during the study. Together, the data hint this ratio could help predict progression.
What about idiopathic disease? Here the data were less clear. In a pilot study of six people with sporadic ALS, the cryptic HDGFL2 was higher than in healthy controls, but the difference missed significance. This may be because the sporadic patients tended to be at a later stage of disease, when cryptic HDGFL2 had already started to fall, Wong noted. He thinks other cryptic peptides might make better markers at this stage.
“More work remains to quantify cryptic exon-containing transcripts and proteins in normal controls, and patients with other diseases such as Alzheimer’s,” noted Johnathan Cooper-Knock at the University of Sheffield, U.K. TDP-43 deposits are found in 40 percent of postmortem AD brains, and isolated TDP-43 pathology, called limbic-predominant age-related TDP-43 encephalopathy neurological change, or LATE-NC, is common in old age (Meneses et al., 2021; Feb 2022 conference news; Mar 2022 conference news). Nelson believes these potential biomarkers should be investigated for this application as well. “LATE-NC is 100-fold more common than ALS, and a context where a biomarker is particularly urgently needed,” he wrote.
If TDP-43 mis-splicing is responsible for neurodegeneration, are there ways to target it therapeutically? A recent paper suggests one approach. In the February 2 Cell Stem Cell, researchers led by Justin Ichida at the University of Southern California, Los Angeles, showed that suppressing the spliceosome-associated factor SYF2 kept cryptic exons out of transcripts in neurons made from both expanded C9ORF72 and sporadic ALS pluripotent stem cells. Suppression of SYF2 also retained TDP-43 in the nucleus and improved neuron health. In mice that overexpress TDP-43, the strategy slowed neurodegeneration.—Madolyn Bowman Rogers
References
News Citations
- Does New Role for ALS-Linked Protein Help Explain Neurodegeneration?
- Beyond the Nucleus: TDP-43 Sticks Together, For Better or Worse
- Microtubule Regulator Connects TDP-43 to Axonal Dysfunction
- Sans Nuclear TDP-43, Splicing of An ALS/FTD Gene Goes Awry
- Virtual Workshop Tackles LATE, a Cause of Late-life Dementia
- Does LATE Subvert Alzheimer's Trials? Biomarkers, Please!
Paper Citations
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011 Oct 20;72(2):245-56. Epub 2011 Sep 21 PubMed.
- Ryan S, Rollinson S, Hobbs E, Pickering-Brown S. C9orf72 dipeptides disrupt the nucleocytoplasmic transport machinery and cause TDP-43 mislocalisation to the cytoplasm. Sci Rep. 2022 Mar 21;12(1):4799. PubMed.
- Halim D, Gao FB. RNA targets of TDP-43: Which one is more important in neurodegeneration?. Transl Neurodegener. 2022 Feb 25;11(1):12. PubMed.
- Offit MB, Wu T, Floeter MK, Lehky TJ. Electrical impedance myography (EIM) in a natural history study of C9ORF72 mutation carriers. Amyotroph Lateral Scler Frontotemporal Degener. 2020 Aug;21(5-6):445-451. Epub 2020 Apr 21 PubMed.
- Sun M, Bell W, LaClair KD, Ling JP, Han H, Kageyama Y, Pletnikova O, Troncoso JC, Wong PC, Chen LL. Cryptic exon incorporation occurs in Alzheimer's brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol. 2017 Jun;133(6):923-931. Epub 2017 Mar 22 PubMed.
- Meneses A, Koga S, O'Leary J, Dickson DW, Bu G, Zhao N. TDP-43 Pathology in Alzheimer's Disease. Mol Neurodegener. 2021 Dec 20;16(1):84. PubMed.
Further Reading
News
- In ALS and FTD, Two Different Routes to TDP-43 Aggregation
- Finally, a Biomarker to Distinguish Primary Tauopathies?
- ALS/FTD Genes Converge on Endolysosomal System, Stoking TDP-43 Pathology
- Liquid or Gel? For TDP-43, the Chaperone HSPB1 Makes the Call
- FTD-GRN: A Disease of Angiogenesis and Vascular Fibrosis?
- Aggregates of TDP-43 in Muscle—Potential ALS Marker?
- Death by Goo: TDP-43 Gels Paralyze Proteasomes in Neurons
Primary Papers
- Seddighi S, Qi YA, Brown AL, Wilkins OG, Bereda C, Belair C, Zhang Y, Prudencio M, Keuss MJ, Khandeshi A, Pickles S, Hill SE, Hawrot J, Ramos DM, Yuan H, Roberts J, Sacramento EK, Shah SI, Nalls MA, Colon-Mercado J, Reyes JF, Ryan VH, Nelson MP, Cook C, Li Z, Screven L, Kwan JY, Shantaraman A, Ping L, Koike Y, Oskarsson B, Staff N, Duong DM, Ahmed A, Secrier M, Ule J, Jacobson S, Rohrer J, Malaspina A, Glass JD, Ori A, Seyfried NT, Maragkakis M, Petrucelli L, Fratta P, Ward ME. Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. bioRxiv. January 23, 2023 bioRxiv
- Irwin KE, Jasin P, Braunstein KE, Sinha I, Bowden KD, Moghekar A, Oh ES, Raitcheva D, Bartlett D, Berry JD, Traynor B, Ling JP, Wong PC. A fluid biomarker reveals loss of TDP-43 splicing repression in pre-symptomatic ALS. bioRxiv. January 24, 2023 bioRxiv
- Linares GR, Li Y, Chang WH, Rubin-Sigler J, Mendonca S, Hong S, Eoh Y, Guo W, Huang YH, Chang J, Tu S, Dorjsuren N, Santana M, Hung ST, Yu J, Perez J, Chickering M, Cheng TY, Huang CC, Lee SJ, Deng HJ, Bach KT, Gray K, Subramanyam V, Rosenfeld J, Alworth SV, Goodarzi H, Ichida J. SYF2 suppression mitigates neurodegeneration in models of diverse forms of ALS. Cell Stem Cell, 2023 February 2 Cell Stem Cell
Follow-On Reading
Papers
- Seddighi S, Qi YA, Brown AL, Wilkins OG, Bereda C, Belair C, Zhang YJ, Prudencio M, Keuss MJ, Khandeshi A, Pickles S, Kargbo-Hill SE, Hawrot J, Ramos DM, Yuan H, Roberts J, Sacramento EK, Shah SI, Nalls MA, Colón-Mercado JM, Reyes JF, Ryan VH, Nelson MP, Cook CN, Li Z, Screven L, Kwan JY, Mehta PR, Zanovello M, Hallegger M, Shantaraman A, Ping L, Koike Y, Oskarsson B, Staff NP, Duong DM, Ahmed A, Secrier M, Ule J, Jacobson S, Reich DS, Rohrer JD, Malaspina A, Dickson DW, Glass JD, Ori A, Seyfried NT, Maragkakis M, Petrucelli L, Fratta P, Ward ME. Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. Sci Transl Med. 2024 Feb 14;16(734):eadg7162. PubMed.
- Irwin KE, Jasin P, Braunstein KE, Sinha IR, Garret MA, Bowden KD, Chang K, Troncoso JC, Moghekar A, Oh ES, Raitcheva D, Bartlett D, Miller T, Berry JD, Traynor BJ, Ling JP, Wong PC. A fluid biomarker reveals loss of TDP-43 splicing repression in presymptomatic ALS-FTD. Nat Med. 2024 Feb;30(2):382-393. Epub 2024 Jan 26 PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
University of Kentucky
This is a remarkable feat of science by a fantastic group of experts from great research centers around the world. It relates to a field of work that has been percolating along for years, and this may represent a significant step forward.
The orientation of the study is toward ALS, which would be interesting and perhaps clinically useful, but the relevance may also exist for LATE-NC, which is 100-fold more common than ALS, and a context where a biomarker is particularly urgently needed.
These studies beget some obvious questions:
I was really impressed by this work by Seddighi and colleagues for several reasons.
First, this is one of the clearest demonstrations that changes at the RNA level in models of disease have substantial and consequential impacts at the protein level.
Second, this work confirms that most TDP-43-related mis-splicing events result in the degradation or instability of affected RNA and proteins. However, in a minority of cases the mis-splicing events create new proteins or proteins with novel sequences that can have unanticipated effects. The authors convincingly show this for one candidate, HDGFL2, and similar findings are likely to affect many other candidates.
Third, while many of the candidates identified in this work may be important for pathogenesis, it is clear that at the very least they will be highly relevant as outcome measures of TDP-43-related function. This, in turn, could be incredibly valuable for the design of fluid biomarkers for patient stratification (for instance, distinguishing FTLD-TDP from FTLD-Tau), target engagement, prognosis and diagnosis.…More
These biomarkers were investigated in CSF, but a more accessible fluid type (i.e. serum) is a possibility that can be pursued in future studies. It's also essential to examine these same markers in ALS/FTD cases vs. controls, and to examine these processes in disorders with predominant TDP-43 pathology (i.e., FTLD-GRN).
Fourth, this is a beautiful demonstration of how cultured human neurons can recapitulate TDP-43 biology seen in vivo, including even in people.
Following up on the seminal discovery that loss of TDP-43 results in the production of cryptic exons, the authors demonstrate that some of these exons can be translated into new polypeptides, thereby altering their interactions with other proteins, and possibly the functions of host proteins.
This interesting finding complements earlier reports that cryptic exons lead to truncation of some full-length host proteins and further supports the notion that correcting multiple TDP-43 targets simultaneously may offer a better chance of success in clinical settings (Halim and Gao, 2022). A major challenge for the future is to identify the subset of TDP-43 targets, if they exist, that may be responsible for the bulk of the consequences of loss of TDP-43 function during disease progression.…More
Equally important is the finding that these de novo polypeptides can be detected in cerebrospinal fluid of patients with ALS, raising the exciting prosect that novel biomarkers can be developed and, if they are sensitive enough, used to monitor TDP-43 pathology in living patients, which would facilitate clinical trials.
References:
Halim D, Gao FB. RNA targets of TDP-43: Which one is more important in neurodegeneration?. Transl Neurodegener. 2022 Feb 25;11(1):12. PubMed.
University of California, San Francisco
I think it is quite an interesting concept to use the expression of aberrant exons as a biomarker for ALS. It potentially takes advantage of the fact that abnormalities in TDP-43 localization are seen in up to 98 percent of ALS patients, and nuclear depletion of TDP-43 is associated with the inclusion of cryptic exons.
A prevailing idea in the ALS field is that abnormalities with TDP-43 are actually pathogenic for ALS, and so a biomarker that is potentially linked to that mechanism could be quite valuable.
University of Sheffield
In the past year the ALS field has been altered by the realization that expression of cryptic exons (CE) under conditions of TDP-43 mis-localization, links together several strands of research, including investigations into ALS-associated genetic variants within UNC13A, which is itself a target of TDP-43 CE splicing. Evidence to date suggests that inclusion of CE within a transcript typically leads to nonsense-mediated decay and reduced expression of the host gene/protein with toxic consequences. However, an interesting question arises over the loss of TDP-43 splicing repression and if it is always pathogenic; after all, as Seddighi and colleagues point out, TDP-43 mis-localization is part of the normal cellular stress response. Another interesting question, which is answered in this preprint, is whether the CE-containing transcripts are ever translated into proteins.…More
This work is primarily based in iPSC-derived neurons, in which the authors have performed CRISPRi knockdown of TDP-43 expression. Under these conditions they were able to confirm expression of CE-containing transcripts and proteins. Given that many stages of cellular quality control can intervene to prevent translation of unwanted proteins, the key step here was the validation, using antibodies and/or shotgun proteomics, that CE-containing proteins are expressed. While this validation was achieved in the iPSC-derived neurons, the authors relied on transcript expression data for postmortem brain tissue. The most exciting part of the study concerned the detection of CE-containing proteins in patient CSF. Here the authors used PRM mass spectrometry proteomics to show that they were able to detect 18 peptides derived from 13 genes in CSF from 15 ALS patients.
Seddighi et al. have developed the CE story by showing that a subset of CE-containing transcripts are actually translated into proteins. The authors propose that these proteins may be toxic—either directly or indirectly via the immune response—or could serve as biomarkers of disease activity, e.g., via detection in CSF. Both of these proposals portend to the question about the normal role of TDP-43 mis-localization in the cellular stress response. A biomarker is most effective if it is disease-specific and more work remains to quantify CE-containing transcripts and proteins in normal controls, and patients with other diseases, such as Alzheimer's. An obvious prediction is that patients with rapidly progressive disease will have more TDP-43 mis-localization, but this analysis was not included in this preprint. The authors state that their assay is subject to technical artefacts, which means that it is only semi-quantitative, but maybe more of these questions will be answered in the final manuscript.
Johns Hopkins
We initially discovered the fundamental role of TDP-43 in repressing non-conserved cryptic exons and suggested that such function is lost in ALS-FTD (Ling et al., 2015); subsequently we also showed this to occur in AD with TDP-43 proteinopathy (Sun et al., 2017) and in Inclusion Body Myositis (Britson et al., 2022).
While recent studies confirm the idea that most of the TDP-43-dependent cryptic exon inclusion would lead to depletion of mRNAs (and their respective proteins), such as STMN2 (Klim et al., 2019; Melamed et al., 2019; Prudencio et al., 2020) and UNC13A (Ma et al., 2022; Brown et al., 2022), this new study by Michael Ward and colleagues focuses on TDP-43 targets with in-frame cryptic exon inclusion (lacking a termination codon), such that the "cryptic" protein can be more easily detected in cells or in biofluid of patients. For example, HDGFL2 is one such TDP-43 target. In fact, we have taken a complementary approach to generate monoclonal antisera directed against this neo-epitope found in HDGFL2. By developing a highly sensitive MSD sandwich ELISA assay, we now show in a large cohort of C9ORF72 patients that loss of TDP-43 splicing repression occurs during early stage disease, including those pre-symptomatic ones.…More
References:
Ling JP, Pletnikova O, Troncoso JC, Wong PC. NEURODEGENERATION. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015 Aug 7;349(6248):650-5. PubMed.
Sun M, Bell W, LaClair KD, Ling JP, Han H, Kageyama Y, Pletnikova O, Troncoso JC, Wong PC, Chen LL. Cryptic exon incorporation occurs in Alzheimer's brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol. 2017 Jun;133(6):923-931. Epub 2017 Mar 22 PubMed.
Britson KA, Ling JP, Braunstein KE, Montagne JM, Kastenschmidt JM, Wilson A, Ikenaga C, Tsao W, Pinal-Fernandez I, Russell KA, Reed N, Mozaffar T, Wagner KR, Ostrow LW, Corse AM, Mammen AL, Villalta SA, Larman HB, Wong PC, Lloyd TE. Loss of TDP-43 function and rimmed vacuoles persist after T cell depletion in a xenograft model of sporadic inclusion body myositis. Sci Transl Med. 2022 Jan 19;14(628):eabi9196. PubMed.
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019 Feb;22(2):167-179. Epub 2019 Jan 14 PubMed.
Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Da Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019 Feb;22(2):180-190. Epub 2019 Jan 14 PubMed.
Prudencio M, Humphrey J, Pickles S, Brown AL, Hill SE, Kachergus JM, Shi J, Heckman MG, Spiegel MR, Cook C, Song Y, Yue M, Daughrity LM, Carlomagno Y, Jansen-West K, de Castro CF, DeTure M, Koga S, Wang YC, Sivakumar P, Bodo C, Candalija A, Talbot K, Selvaraj BT, Burr K, Chandran S, Newcombe J, Lashley T, Hubbard I, Catalano D, Kim D, Propp N, Fennessey S, NYGC ALS Consortium, Fagegaltier D, Phatnani H, Secrier M, Fisher EM, Oskarsson B, van Blitterswijk M, Rademakers R, Graff-Radford NR, Boeve BF, Knopman DS, Petersen RC, Josephs KA, Thompson EA, Raj T, Ward M, Dickson DW, Gendron TF, Fratta P, Petrucelli L. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest. 2020 Nov 2;130(11):6080-6092. PubMed.
Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, Briner A, Rodriguez CM, Guo C, Akiyama T, Schmidt HB, Cummings BB, Wyatt DW, Kurylo K, Miller G, Mekhoubad S, Sallee N, Mekonnen G, Ganser L, Rubien JD, Jansen-West K, Cook CN, Pickles S, Oskarsson B, Graff-Radford NR, Boeve BF, Knopman DS, Petersen RC, Dickson DW, Shorter J, Myong S, Green EM, Seeley WW, Petrucelli L, Gitler AD. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature. 2022 Mar;603(7899):124-130. Epub 2022 Feb 23 PubMed.
Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, Bampton A, Lee FC, Masino L, Qi YA, Bryce-Smith S, Gatt A, Hallegger M, Fagegaltier D, Phatnani H, NYGC ALS Consortium, Newcombe J, Gustavsson EK, Seddighi S, Reyes JF, Coon SL, Ramos D, Schiavo G, Fisher EM, Raj T, Secrier M, Lashley T, Ule J, Buratti E, Humphrey J, Ward ME, Fratta P. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. 2022 Mar;603(7899):131-137. Epub 2022 Feb 23 PubMed. Correction.
University of Toronto
This is great work; however, it is not the first time proteins derived from cryptic exons have been described in ALS. We reported a peripherin splice variant retaining intron 4 and encoding Per 61 protein in mutant SOD1 mice (Robertson et al., 2003), and Per 28 retaining introns 3 and 4 in ALS tissues (Xiao et al., 2008). We also suggested that these isoforms could be used as biomarkers to differentiate between disease/injury paradigms (McLean et al., 2010).
References:
Robertson J, Doroudchi MM, Nguyen MD, Durham HD, Strong MJ, Shaw G, Julien JP, Mushynski WE. A neurotoxic peripherin splice variant in a mouse model of ALS. J Cell Biol. 2003 Mar 17;160(6):939-49. PubMed.
Xiao S, Tjostheim S, Sanelli T, McLean JR, Horne P, Fan Y, Ravits J, Strong MJ, Robertson J. An aggregate-inducing peripherin isoform generated through intron retention is upregulated in amyotrophic lateral sclerosis and associated with disease pathology. J Neurosci. 2008 Feb 20;28(8):1833-40. PubMed. …More
McLean J, Liu HN, Miletic D, Weng YC, Rogaeva E, Zinman L, Kriz J, Robertson J. Distinct biochemical signatures characterize peripherin isoform expression in both traumatic neuronal injury and motor neuron disease. J Neurochem. 2010 Aug;114(4):1177-92. PubMed.
University of Auckland
The University of Auckland
These two preprints beautifully outline the potential for RNA splicing changes related to TDP-43 proteinopathy to be harnessed as biomarkers, through their translation into more readily tested cryptic peptides. This represents a significant conceptual leap, analogous to the translation of the intronic C9orf72 repeat expansion into dipeptide repeats.
Taken together, Seddighi et al. and Irwin et al. present compelling evidence that the cryptic peptide of HDGFL2 can distinguish C9orf72-linked cases from controls and predict symptom onset, particularly when combined with neurofilament H or L. It will be of great interest to see how this biomarker performs in larger cohorts of sporadic cases and controls. Combining a panel of these novel cryptic peptide antibodies may improve case/control discrimination against heterogeneous genetic and clinical backgrounds.…More
In our own preprint, deposited this week (Cao et al., 2023), we depleted TARDBP from human brain-derived pericytes, generating one of the few non-reprogrammed human TDP-43 loss-of-function models. Combined with previous studies (Cao and Scotter, 2022; Brown et al., 2022; Klim et al., 2019; Ling et al., 2015; Ma et al., 2022; Melamed et al., 2019; Shiga et al., 2012), we identified TDP-43-related splicing changes in human ALS motor cortex tissue. Indeed, combining a panel of such splicing changes discriminated ALS tissue from controls.
Splicing changes and cryptic peptides are probably more specific and proximal reporters of TDP-43 loss of function than differential gene expression, and the detection of cryptic peptides in the CSF is hugely exciting. We look forward to the application of these findings to better understanding TDP-43 dysfunction, including in non-neuronal cell types, and to improving therapeutic options for people living with ALS and FTD.
References:
Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, Bampton A, Lee FC, Masino L, Qi YA, Bryce-Smith S, Gatt A, Hallegger M, Fagegaltier D, Phatnani H, NYGC ALS Consortium, Newcombe J, Gustavsson EK, Seddighi S, Reyes JF, Coon SL, Ramos D, Schiavo G, Fisher EM, Raj T, Secrier M, Lashley T, Ule J, Buratti E, Humphrey J, Ward ME, Fratta P. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. 2022 Mar;603(7899):131-137. Epub 2022 Feb 23 PubMed. Correction.
Cao MC, Scotter EL. Transcriptional targets of amyotrophic lateral sclerosis/frontotemporal dementia protein TDP-43 - meta-analysis and interactive graphical database. Dis Model Mech. 2022 Sep 1;15(9) Epub 2022 Sep 13 PubMed.
Cao MC, Ryan B, Wu J, Curtis MA, Faull RL, Dragunow M, Scotter EL. Differential exon usage and cryptic exon profiles of TDP-43 loss of function in amyotrophic lateral sclerosis brain tissue. bioRxiv. February 4, 2023 bioRxiv
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019 Feb;22(2):167-179. Epub 2019 Jan 14 PubMed.
Ling JP, Pletnikova O, Troncoso JC, Wong PC. NEURODEGENERATION. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015 Aug 7;349(6248):650-5. PubMed.
Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, Briner A, Rodriguez CM, Guo C, Akiyama T, Schmidt HB, Cummings BB, Wyatt DW, Kurylo K, Miller G, Mekhoubad S, Sallee N, Mekonnen G, Ganser L, Rubien JD, Jansen-West K, Cook CN, Pickles S, Oskarsson B, Graff-Radford NR, Boeve BF, Knopman DS, Petersen RC, Dickson DW, Shorter J, Myong S, Green EM, Seeley WW, Petrucelli L, Gitler AD. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature. 2022 Mar;603(7899):124-130. Epub 2022 Feb 23 PubMed.
Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Da Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019 Feb;22(2):180-190. Epub 2019 Jan 14 PubMed.
Shiga A, Ishihara T, Miyashita A, Kuwabara M, Kato T, Watanabe N, Yamahira A, Kondo C, Yokoseki A, Takahashi M, Kuwano R, Kakita A, Nishizawa M, Takahashi H, Onodera O. Alteration of POLDIP3 splicing associated with loss of function of TDP-43 in tissues affected with ALS. PLoS One. 2012;7(8):e43120. PubMed.
View all comments by Maize CaoPenn State College of Medicine
Using sophisticated, highly sensitive, albeit different approaches, these two papers show that “cryptic” protein sequences can be detected in CSF from ALS/FTD patients. These elusive protein sequences have long been predicted to exist by Dr. Phil Wong and others in the field; however, their presence has only now been demonstrated, for the first time.
These elegant new detection methods will allow researchers and clinicians to better predict disease progression, which is especially important for asymptomatic mutation carriers, and to better evaluate the outcomes of clinical trials. While the presence of “cryptic” protein sequences reveals an additional layer of complexity in the pathophysiology of ALS/FTD, the real game-changer here lies in the ability of the ”cryptic” protein sequences to serve as much-awaited and much-needed biomarkers for ALS/FTD.…More
Make a Comment
To make a comment you must login or register.