Liquid or Gel? For TDP-43, the Chaperone HSPB1 Makes the Call
Quick Links
In most people with ALS, and many with FTD, cytoplasmic aggregates of TDP-43 plague degenerating neurons. What causes an RNA-binding protein that spends most of its time in the nucleus to pile up in the cytoplasm? According to work published September 8 in Nature Cell Biology, TDP-43 could get into cytoplasmic mischief in the absence of a small heat shock protein called HSPB1. Scientists led by Don Cleveland at the University of California, San Diego, found that under conditions of cellular stress, HSPB1 ushers TDP-43 in the cytoplasm into liquid droplets there. The chaperone maintains TDP-43 in this fluid state, keeping the droplets from hardening into gels and thus preventing formation of TDP-43 fibrils. Then, after stress passes, HSPB1 teams up with other chaperones to disassemble the droplets. In people with ALS, motor neurons with cytoplasmic TDP-43 aggregates had scant HSPB1. In all, the findings suggest that HSPB1 helps preserve the fluidity of TDP-43 in the cytoplasm, sparing it from permanent entrapment within aggregates there.
- In response to stress, HSPB1 ushers TDP-43 into liquid droplets in the cytoplasm.
- HSPB1 keeps droplets from hardening into gels, and blocks TDP-43 fibril formation.
- Motor neurons from people with ALS have too little HSPB1.
“This exciting study reports that in cultured cells, the small heat shock protein HSPB1 regulates the phase separation behavior of cytoplasmic TDP-43,” wrote Ludo Van Den Bosch of KU Leuven in Belgium. “That the formation of these TDP-43 droplets leads to the depletion of nuclear TDP-43 indicates that what is observed in a cell line reflects what happens in patients.”
Previously, scientists in Cleveland’s lab had pegged chaperones, chiefly HSP70, as being responsible for regulating the formation of TDP-43-containing droplets in the nucleus. Called anisosomes, these intranuclear droplets formed in response to proteosome inhibition, or when TDP-43 was rendered unable to bind to RNA (Yu et al., 2021). The current work investigates how phase separation of TDP-43 is controlled within the cytoplasm.
To explore TDP-43 phase separation in the cytoplasm, first author Shan Lu and colleagues inducibly expressed a version of TDP-43 sans its nuclear localization sequence in U2OS cells. Inducing stress in these cells—either by blocking the proteosome or by activating oxidative stress—triggered TDP-43 to separate, or “de-mix,” into liquid droplets. At first, these droplets changed shape with ease as they moved freely throughout the cytoplasm. After a few hours of stress, these free-wheeling droplets had coalesced and hardened into stubborn gels. The researchers ultimately pegged TDP-43’s RRM1, an RNA-binding protein domain, as the facilitator of this liquid-to-gel transition.
Even in the absence of acute stressors, cells expressing cytoplasmic TDP-43 hosted a mix of small liquid droplets and larger, elongated gels containing the protein, which were distinct from stress granules. The cytoplasmic droplets managed to recruit and ensnare full-length, nuclear TDP-43, ultimately depriving the nucleus of it and its function.
What controls this balance of TDP-43 phase separation in the cytoplasm? To address this question, Lu looked at which proteins TDP-43 buddied up with there. A proximity labeling technique called APEX2, coupled with mass spectrometry, dredged up 15 proteins that directly interact with TDP-43 in the cytoplasm. Only HSPB1 tripled its interaction with TDP-43 under conditions of oxidative stress. The scientists also spotted HSPB1 mingling with TDP-43 in cytoplasmic droplets in iPSC-derived cortical and motor neurons, supporting the idea that it has a droplet-specific relationship with TDP-43 in neurons.
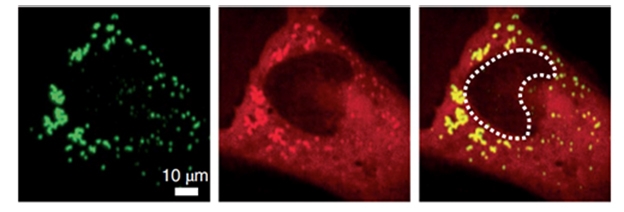
De-Mix Together. When there is oxidative stress, TDP-43 (green) and HSPB1 (red) co-segregate into liquid droplets within the cytoplasm (yellow). [Courtesy of Lu et al., Nature Cell Biology, 2022.]
By changing HSPB1 and TDP-43’s concentrations in vitro, the researchers found that the former ushered the latter into liquid droplets, but prevented the droplets from hardening into gels or solids. The chaperone also blocked TDP-43 from twisting into amyloid fibrils. In cells, most of the TDP-43-containing liquid droplets dissipated after the stressor—in this case, sodium arsenite—was removed. The researchers also report that HSPB1 helped orchestrate the disassembly of the droplets along with a handful of other chaperones, including HSP70, BAG2, and HSPA1A.

HSPB1 To TDP-43: Don’t Gel! With sufficient HSPB1 present in a situation of stress (left), the chaperone whisks TDP-43 into droplets, then coordinates with other chaperones to disassemble the droplets later. When HSPB1 is scarce (right), cytoplasmic TDP-43 de-mixes into droplets and forms gels and fibrils. [Courtesy of Lu et al., Nature Cell Biology, 2022]
Might a dearth of HSPB1 contribute to the accumulation of cytoplasmic TDP-43 aggregates in ALS? The scientists measured levels of HSPB1 and TDP-43 pathology in motor neurons from spinal cords of people with or without the disease. They found that motor neurons from patients had less HSPB1 than those from controls. Among people with ALS, motor neurons with cytoplasmic TDP-43 aggregates had less HSPB1 than motor neurons without that pathology. In further support of a potential role for HSPB1 in ALS, the researchers found an enrichment of missense and frameshift variants in the HSPB1 gene in people with the disease relative to controls.
The findings extend studies in the aughts, which had identified genetic variants in HSPB1 in people with ALS, implicating the small heat shock protein in that disease (Dierick et al., 2007). Scientists led by van den Bosch soon found that overexpressing HSPB1 in a transgenic mouse model of ALS caused by SOD1-G93A mutation failed to rescue the mice from the disease, despite reports that HSPB1 also bound to SOD1 (Krishnan et al., 2008).
“Crossbreeding a TDP-43 mouse model with these HSPB1-overexpressing mice seems to be a logical next step,” Van Den Bosch commented to Alzforum. He noted that the study raises the important question of whether insights about HSPB1 can be translated into a therapeutic strategy for ALS. Riling the heat shock response was the rationale behind a recently failed Phase 3 trial (NCT03491462), he added. “Overall, this interesting study renews the interest into chaperones in general and HSPB1 in particular,” he wrote.
“The Cleveland group has nicely shown that HSPB1 regulates TDP-43 phase separation and aggregate formation,” commented Benjamin Wolozin of Boston University in Massachusetts (full comment below).” “Importantly they show that modulating levels of HSPB1 is capable of regulating phase separation of TDP-43; this finding is important because chaperones able to modulate TDP-43 phase separation become potential targets for disease modification.”
Variants in HSPB1 have also been tied to Charcot-Marie-Tooth disease, an inherited neuromuscular disease (Evgrafov et al., 2004). Sami Barmada of University of Michigan in Ann Arbor noted that mutations associated with CMT increase HSPB1 chaperone activity. “It will be really interesting to determine if the same holds true for ALS-associated mutations, rather than a loss of function as suggested by the authors. If correct, this would argue against a toxic contribution by stable, i.e., gel or fibril-like, inclusions in ALS,” he wrote.—Jessica Shugart
References
Paper Citations
- Yu H, Lu S, Gasior K, Singh D, Vazquez-Sanchez S, Tapia O, Toprani D, Beccari MS, Yates JR 3rd, Da Cruz S, Newby JM, Lafarga M, Gladfelter AS, Villa E, Cleveland DW. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science. 2021 Feb 5;371(6529) Epub 2020 Dec 17 PubMed.
- Dierick I, Irobi J, Janssens S, Theuns J, Lemmens R, Jacobs A, Corsmit E, Hersmus N, Van Den Bosch L, Robberecht W, De Jonghe P, Van Broeckhoven C, Timmerman V. Genetic variant in the HSPB1 promoter region impairs the HSP27 stress response. Hum Mutat. 2007 Aug;28(8):830. PubMed.
- Krishnan J, Vannuvel K, Andries M, Waelkens E, Robberecht W, Van Den Bosch L. Over-expression of Hsp27 does not influence disease in the mutant SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Neurochem. 2008 Sep;106(5):2170-83. PubMed.
- Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, Schagina O, Verpoorten N, Van Impe K, Fedotov V, Dadali E, Auer-Grumbach M, Windpassinger C, Wagner K, Mitrovic Z, Hilton-Jones D, Talbot K, Martin JJ, Vasserman N, Tverskaya S, Polyakov A, Liem RK, Gettemans J, Robberecht W, De Jonghe P, Timmerman V. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004 Jun;36(6):602-6. Epub 2004 May 2 PubMed.
Further Reading
Papers
- Krishnan J, Vannuvel K, Andries M, Waelkens E, Robberecht W, Van Den Bosch L. Over-expression of Hsp27 does not influence disease in the mutant SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Neurochem. 2008 Sep;106(5):2170-83. PubMed.
Primary Papers
- Lu S, Hu J, Arogundade OA, Goginashvili A, Vazquez-Sanchez S, Diedrich JK, Gu J, Blum J, Oung S, Ye Q, Yu H, Ravits J, Liu C, Yates JR 3rd, Cleveland DW. Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat Cell Biol. 2022 Sep;24(9):1378-1393. Epub 2022 Sep 8 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Neurobiology, KU Leuven & VIB
This exciting study by the research group of Dr. Cleveland reports that in cultured cells the small heat shock protein HSPB1 regulates the phase separation behavior of cytoplasmic TDP-43. Interestingly, TDP-43 phase separates independently from stress granules without the requirement to bind RNA. The fact that the formation of these TDP-43 droplets leads to the depletion of nuclear TDP-43 indicates that what is observed in a cell line reflects what happens in patients. Another indication is that the expression level of HSPB1 in postmortem tissue of ALS patients is decreased in those spinal motor neurons with TDP-43 pathology. Remarkably, HSPB1 does not only interact with TDP-43 (and FUS) but also with SOD1 which inspired us several years ago to overexpress HSPB1 in the mutant SOD1 mouse model. Unfortunately without any benefit (Krishnan et al., 2008). Crossbreeding a TDP-43 mouse model with these HSPB1 overexpressing mice seems to be a logical next step. This brings us to the important question whether these insights can be translated into a therapeutic strategy beneficial for ALS patients. Upregulating the heat-shock response was the rationale to test arimoclomol in ALS (Kieran et al., 2004), which unfortunately failed in a Phase 3 trial (NCT03491462). Overall, this interesting study renews the interest into chaperones in general and HSPB1 in particular. In addition, the interaction of TDP-43 with tubulins and the requirement for intact microtubules to get TDP-43 droplet disassembly further increases the excitement.…More
References:
Krishnan J, Vannuvel K, Andries M, Waelkens E, Robberecht W, Van Den Bosch L. Over-expression of Hsp27 does not influence disease in the mutant SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Neurochem. 2008 Sep;106(5):2170-83. PubMed.
Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004 Apr;10(4):402-5. PubMed.
Boston University School of Medicine
This is supremely well-designed work from the Cleveland lab. As mentioned in the article, many proteins linked to proteostasis exhibit genetic linkage to ALS. In addition, chaperones and other proteins linked to proteostasis frequently show up in molecular studies of TDP-43 protein interaction networks, as well as interaction networks of other RNA-binding proteins. Thus, the observed role of HSPB1 is not surprising because biomolecular condensates and subsequent fibrillar aggregates are associated with lots of these proteins in chaperones and other proteins regulating proteostasis.
However, just because something is unsurprising does not mean that it is not important. The Cleveland group has nicely shown that HSPB1 regulates TDP-43 phase separation and aggregate formation. Importantly they show that modulating levels of HSPB1 is capable of regulating phase separation of TDP-43; this finding is important because chaperones able to modulate TDP-43 phase separation become potential targets for disease modification, i.e., therapy.…More
The challenge is to find those interacting proteins that exhibit the strongest effects on TDP-43 phase separation. HSPB1 clearly has strong effects, particularly when one examines resolution of TDP-43 biomolecular condensates at 12 hours, where siHSPB1 prevents removal of the phase separated TDP-43 droplets (Fig. 7g).
Three other points:
1) The article shows a nice correlation between cytoplasmic TDP-43 phase separation and loss of nuclear TDP-43. Such nuclear loss presumably lies behind the extensive cryptic splicing observed by Phil Wong, Len Petrucelli, Aaron Gitler, Michael Ward and others.
2) I note the absence of co-localization between G3BP1 and TDP-43. The Cleveland group has reported this previously and suggest that TDP-43 granules are something different than the classic stress granule. There are many types of RNA granules, which might respond to differing signaling pathways. The absence of G3BP1–TDP-43 co-localization speaks to this heterogeneity.
3) The work is overwhelmingly done in non-neuronal cells that are easy to use, but the Cleveland group does a nice job of showing that these findings translate to motor neurons, which is always a key thing to look for in such studies.
In general, this is a beautiful study of TDP43 variant dynamics in vitro and in cultured cells, focusing on the changes in cytoplasmic TDP43 mobility upon cellular stress. One of the most important findings is that HSPB1 acts as a chaperone for TDP43, regulating its biophysical state as well as its localization in cells. Consistent with this function, HSPB1 is lost from cells that exhibit TDP43 pathology (nuclear exclusion, cytoplasmic deposition) in ALS. Loss of HSPB1 is also associated with a more pervasive interruption of nucleocytoplasmic transport, with several typically nuclear proteins becoming mislocalized, not just TDP43.
This includes RanGAP, a factor critical for regulating Ran GDP/GTP balance required for transport. Notably, HSPB1 is highly expressed in motor neurons, and mutations in the HSPB1 gene have been associated with ALS and other conditions that affect motor neurons, including subtypes of Charcot-Marie-Tooth disease. These observations support a fundamental connection between HSPB1, TDP43 and motor neuron health.…More
Keeping all this in mind, it is unclear how relevant the cellular stress paradigms utilized in this work (MG132, NaAsO2, heat) are for reproducing the ALS disease state. In fact, more recent work suggests that, if anything, heat stress may be the most similar on a molecular basis, even if not an intuitive basis. Likewise, many of the manipulations utilized here could be unrelated to disease. These include transient overexpression of dNLS TDP43, and/or dNLS TDP43 carrying mutations that mimic acetylation. Most experiments were performed in U2OS cells, a transformed cell line that may exhibit unique responses to disease-related stimuli such as TDP43 accumulation.
On a different and thought-provoking note, prior evidence suggests that disease-associated (CMT) mutations in the HSPB1 gene increase chaperone-like activity. It will be really interesting to determine if the same holds true for ALS-associated mutations, rather than a loss of function as suggested by the authors (a "probable" enrichment for missense or frame-shift variants in ALS, p=0.068). If correct, this would argue against a toxic contribution by stable (i.e. gel or fibril-like) inclusions in ALS.
Make a Comment
To make a comment you must login or register.