In ALS and FTD, Two Different Routes to TDP-43 Aggregation
Quick Links
Different genetic mutations can converge on the same downstream pathology. The C9ORF72 repeat expansion and progranulin haploinsufficiency both trigger the RNA-binding protein TDP-43 to abandon the nucleus and settle in cytoplasm. TDP-43 deposits underlie cases of frontotemporal dementia and amyotrophic lateral sclerosis. How does each mutation promote this pathology? Quite differently, according to two new papers.
- Poly(GR) made from the C9ORF72 repeat expansion sequesters TDP-43.
- Antisense oligonucleotides suppress poly(GR) and prevent this pathology.
- Complement proteins released by progranulin-deficient microglia induce TDP-43 aggregation in neurons.
In the September 2 Science Translational Medicine, researchers led by Leonard Petrucelli and Yong-Jie Zhang at the Mayo Clinic in Jacksonville, Florida, and James Shorter at the University of Pennsylvania, Philadelphia, lay the blame on the dipeptide repeat protein poly-glycine-arginine (GR), which results from aberrant translation of C9ORF72 repeat expansion transcripts. The researchers report that poly(GR) directly interacts with TDP-43, pulling it into aggregates. In a mouse model, suppressing poly(GR) production with antisense oligonucleotides prevented TDP-43 aggregation and subsequent neurodegeneration.
“This impressive new paper from the Petrucelli group presents a unified hypothesis linking key molecular phenotypes of C9ORF72-ALS to a single mechanism,” Johnathan Cooper-Knock at the University of Sheffield, U.K., wrote to Alzforum (full comment below). “The researchers have shown that poly(GR) is sufficient to reproduce all of the important aspects of TDP-43 pathology seen in human patients.”
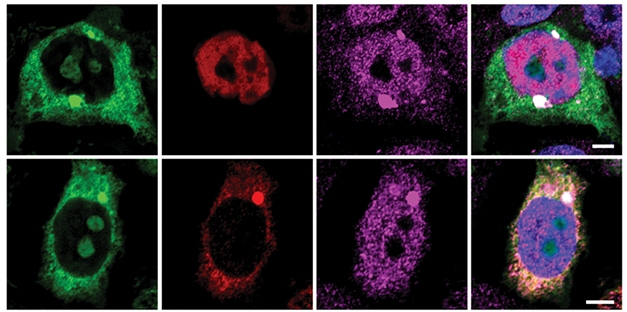
Coaxing TDP-43. In cells with wild-type TDP-43 (red), the protein remains nuclear (top row). When forced into cytoplasm (bottom row), TDP-43 aggregated with poly(GR) (green) and drew the stress granule protein TIA-1 (purple) into these deposits as well. [Courtesy of Cook et al., Science Translational Medicine.]
But what about cases of TDP-43 proteinopathy with other genetic causes? In the August 31 Nature, researchers led by Eric Huang at the University of California, San Francisco, focused on FTD cases caused by loss-of-function mutations in the secreted growth factor progranulin. In a mouse model, they found that a dearth of progranulin affected microglia more than any other cell in the brain. In its absence, microglia release more complement proteins. These innate immune factors in turn damage excitatory neurons, causing TDP-43 to accumulate in cytoplasm and setting the stage for its aggregation there. “Microglia are the major factor driving disease in FTD cases caused by progranulin mutation,” Huang concluded.
Rogue Polypeptides Stir Up Trouble
Aberrant translation of the C9ORF72 repeat expansion results in the production of several polydipeptide proteins, including poly-glycine-alanine (GA) and poly(GR). A previous study found that expressing these proteins in flies was sufficient to draw TDP-43 out of the nucleus and into cytoplasm, where it formed aggregates (Oct 2018 news). Petrucelli and colleagues had generated a mouse model that expressed the C9ORF72 repeat expansion and accumulated cytoplasmic TDP-43 deposits (Chew et al., 2015). However, it was unclear how the aberrant polypeptides caused this.
To investigate, co-first author Hana Odeh at UPenn combined recombinant TDP-43 with either poly(GA) or poly(GR) in a cell-free assay. Poly(GA) had no effect on TDP-43, but poly(GR) accelerated its aggregation, leading to dense deposits. Co-first authors Casey Cook and Yanwei Wu at the Mayo Clinic followed this up by transfecting a kidney cell line with each polypeptide and TDP-43. The TDP-43 construct lacked a nuclear localization signal, keeping it cytoplasmic. Again, poly(GR) aggregated with TDP-43, but poly(GA) did not (see image above). By mutating various regions of TDP-43, the authors determined that its ability to bind poly(GR) did not depend on its RNA-binding motifs, nor on its sticky C-terminal tail. Ongoing work suggests that the N-terminal domain of TDP-43 is responsible for binding poly(GR), Petrucelli told Alzforum.
The authors moved to mice, injecting a 200-repeat poly(GR) construct into the lateral ventricles of newborns. After two weeks, about 8 percent of transfected cortical cells formed polydipeptide aggregates. These almost always contained TDP-43 as well, along with stress granule markers such as ataxin-2. “Stress granules appear to be recruited to the multimeric poly(GR)-TDP-43 complex and perhaps stabilize the structure, forming an inclusion,” Petrucelli said. In addition, the aggregates sequestered nucleoporins and nucleocytoplasmic transport factors, including importin α5 and karyopherin α2. Several previous studies have found that dipeptide repeat proteins jam up nuclear traffic by depleting these factors (Jan 2018 news; Nov 2019 news). By contrast, in mice that expressed poly(GA), polydipeptide repeat inclusions formed, but did not recruit TDP-43, or other factors.
Would suppressing poly(GR) production prevent TDP-43 aggregation? The authors turned to a mouse model that expresses a C9ORF72 expansion with 149 repeats. These animals make all the polypeptides and develop TDP-43 aggregates and neurodegeneration as they age. The researchers injected C9 antisense oligonucleotides (ASOs) into the brains of 3-month-old mice, which have abundant polydipeptide repeat deposits but no TDP-43 mislocalization yet. Three months later, treated animals had far fewer deposits containing TDP-43 than did untreated littermates. Untreated mice lost neurons over this time period, and the neurodegeneration marker neurofilament light shot up in the plasma. ASO treatment maintained neurons and plasma NfL at wild-type levels.
That antisense oligonucleotide treatment can ameliorate TDP-43 pathology is new, Petrucelli noted. “That’s a message of optimism for trials targeting C9ORF72 RNA,” he said. He believes the finding will translate to the clinic, because he found poly(GR) and TDP-43 sequestering together in postmortem tissue samples from people with the C9ORF72 expansion. ASOs directed against C9ORF72 are in clinical trials (May 2018 conference news).
“This paper describes very exciting results,” Brian Freibaum at St. Jude Children’s Research Hospital in Memphis, Tennessee, wrote to Alzforum (full comment below). “I would be interested to know if the interaction between TDP-43 and poly(GR) could be inhibited by small molecules or peptides and if so, whether this would rescue poly(GR)-mediated toxicity.”
Robert Baloh at Cedars-Sinai Medical Center, New York, agreed that the evidence suggests poly(GR) drives TDP-43 pathology. “Several interesting questions remain for further exploration, such as what drives TDP-43 pathology in non-C9ORF72 cases of FTD/ALS where dipeptide repeat proteins are not present,” he wrote to Alzforum (full comment below).
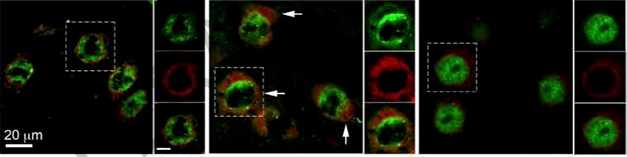
Complement Lures TDP-43 Away. In neurons from wild-type mice (left), TDP-43 (green) is nuclear, and nucleoporin Nup98 (red) stays in the nuclear envelope. In GRN knockouts (center), both escape into cytoplasm, but when the knockouts also lack C1q and C3 (right), the proteins stay put. [Courtesy of Zhang et al., Nature.]
Microglia Play the Heavy in Progranulin Disease
A case in point are FTD cases caused by progranulin mutations. Progranulin participates in lysosomal processing, the stress response, and innate immunity (for review see Kao et al., 2017). Huang and colleagues previously found that low levels of progranulin cause microglia to supercharge production of complement proteins, leading to synapse elimination (May 2016 news).
To look more closely at how loss of progranulin affects microglia and other cell types, joint first authors Jiasheng Zhang, Dmitry Velmeshev, Kei Hashimoto, and Yu-Hsin Huang used RNA-Seq to compare gene expression in the thalami of wild-type and GRN knockout mice at 2, 4, 7, 12, and 19 months of age. The thalamus typically atrophies in FTD (Bocchetta et al., 2018). In wild-type animals, microglia express much more progranulin than do other cell types. Perhaps not surprisingly, then, thalamic microglia were the first cell type to show effects in the knockouts. At 7 months of age, expression of 45 microglial genes either rose or fell, and this went up to 65 genes at 12 months and 265 at 19 months. In general, microglia turned down homeostatic genes and boosted inflammatory and lysosomal genes. The gene expression profile partially overlapped with microglial changes seen in Alzheimer’s disease and ALS, the authors noted (Jun 2017 news; Sep 2017 news).
The microglial changes had their greatest effect on excitatory neurons in the thalamus. At 12 months, these neurons accumulated cytoplasmic TDP-43. By 19 months, some of the neurons had died off, while gene expression in the survivors had gone haywire.
To parse what was going on between microglia and neurons, the authors established primary cell cultures of each from wild-type and knockout mice. Cultured microglia are notorious for changing their phenotype, but by using serum-free culture conditions, Huang and colleagues were able to maintain these cells in something close to their native state, with a similar gene expression profile to microglia in mouse brain (Jun 2017 news). After 14 days in vitro, the authors collected media from the microglial cultures and added them to the excitatory neuron cultures. They found that conditioned media from knockout microglia was far more toxic than that from wild-types. It killed 30 percent of wild-type excitatory neurons, and more than half of GRN knockout neurons. Curiously, inhibitory neurons were fairly resistant to these toxic effects.
How did conditioned media kill? When added to GRN-/- neurons, it depleted nuclear pore proteins and caused TDP-43 to bolt into the cytoplasm. Cytoplasmic TDP-43 partially localized with lysosomes but, unlike in Petrucelli’s study, attracted no stress granules. Because the authors previously found that progranulin knockout microglia make more C1q and C3b complement protein than do wild-types, they wondered if these inflammatory factors might mediate the harmful effects. Indeed, treating cultured wild-type and GRN-/- neurons with C1q, with or without C3b, mimicked the effects of conditioned media and triggered cytoplasmic TDP-43 granules. However, it did not kill as many neurons as did conditioned media, suggesting there might be other toxic factors involved.
However, deleting C1q or C3 from knockout microglia rendered their conditioned media nontoxic, suggesting these factors are essential for toxicity. Vitronectin, which prevents complement proteins from forming a membrane attack complex, likewise protected neurons from the toxic effects of microglial conditioned media. Progranulin knockout mice lacking C1q and C3 did not accumulate TDP-43, and their neurons stayed healthy (see image above).
Huang is now analyzing gene expression in postmortem human brain from progranulin carriers to see if similar changes occur as in mice. If so, it would suggest that microglia are the cell type to target in FTD caused by progranulin mutations, Huang said. Such therapies should aim to cool microglial activation, he suggested.
Luke Daly at the University of Massachusetts Medical School, Worcester, agreed. “These findings further elevate the complement system as a potentially viable therapeutic target for PGRN-mediated FTD,” he wrote to Alzforum (full comment below). Shorter was intrigued by the fact that the data appear to describe a path to cytoplasmic TDP-43 aggregation that is independent of stress granules.
These findings further imply that complement proteins could become a biomarker of disease progression in progranulin mutation carriers. On this, Huang’s preliminary data show that complement in cerebrospinal fluid rises as progranulin carriers become impaired. “Perhaps CSF complement level could be used as an indicator for treatment efficacy,” he speculated.—Madolyn Bowman Rogers
References
News Citations
- Dipeptide Repeat Proteins Trigger TDP-43 Pathology, Faulty Nuclear Import
- TDP-43 Snarls Nuclear Traffic
- Dipeptide Repeat Proteins Don’t Directly Block Nuclear Transport
- At AAN, Sights Set on Antisense Therapies for Diseases of the Brain
- Microglia Prune Synapses in a Subtype of Frontotemporal Dementia
- Hot DAM: Specific Microglia Engulf Plaques
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- What Makes a Microglia? Tales from the Transcriptome
Paper Citations
- Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A, Overstreet K, Edbauer D, Rademakers R, Boylan KB, Dickson DW, Fryer JD, Petrucelli L. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015 Jun 5;348(6239):1151-4. Epub 2015 May 14 PubMed.
- Kao AW, McKay A, Singh PP, Brunet A, Huang EJ. Progranulin, lysosomal regulation and neurodegenerative disease. Nat Rev Neurosci. 2017 Jun;18(6):325-333. Epub 2017 Apr 24 PubMed.
- Bocchetta M, Gordon E, Cardoso MJ, Modat M, Ourselin S, Warren JD, Rohrer JD. Thalamic atrophy in frontotemporal dementia - Not just a C9orf72 problem. Neuroimage Clin. 2018;18:675-681. Epub 2018 Feb 23 PubMed.
Further Reading
News
- C9ORF72 Toxicity Tied to Mitochondria, Transcriptional Machinery
- Beyond the Nucleus: TDP-43 Sticks Together, For Better or Worse
- It’s ‘And,’ Not ‘Either-Or’: C9ORF72 Mechanisms of Action are Linked
- In New ALS/FTD Mouse Model, Poly(GR) Peptides Poison Ribosomes
- Dipeptide Repeats May Hobble Ribosomes in C9ORF72-FTD Patient Brain
- No, TDP-43 and FUS Are Not Actively Exported From the Nucleus
- Lack of C9ORF72 Protein Renders Neurons More Vulnerable to Degeneration
- Stressed-Out Cells Translate C9ORF72 Repeats, Unleash Toxic Peptides
- Stabilizing C9ORF72 RNA Lowers Toxicity
- Granulins: The Missing Link between Progranulin and Lysosome Function?
Primary Papers
- Cook CN, Wu Y, Odeh HM, Gendron TF, Jansen-West K, Del Rosso G, Yue M, Jiang P, Gomes E, Tong J, Daughrity LM, Avendano NM, Castanedes-Casey M, Shao W, Oskarsson B, Tomassy GS, McCampbell A, Rigo F, Dickson DW, Shorter J, Zhang YJ, Petrucelli L. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci Transl Med. 2020 Sep 2;12(559) PubMed.
- Zhang J, Velmeshev D, Hashimoto K, Huang YH, Hofmann JW, Shi X, Chen J, Leidal AM, Dishart JG, Cahill MK, Kelley KW, Liddelow SA, Seeley WW, Miller BL, Walther TC, Farese RV Jr, Taylor JP, Ullian EM, Huang B, Debnath J, Wittmann T, Kriegstein AR, Huang EJ. Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature. 2020 Dec;588(7838):459-465. Epub 2020 Aug 31 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Director of Neuromuscular Medicine, Cedars-Sinai Medical Center
The origin and meaning of TDP-43 aggregation in neurodegenerative disorders such as ALS and FTD remain a mystery. TDP-43 undergoes physiology aggregation and disaggregation, and, as an RNA-binding protein, traverses in and out of the nucleus. Somehow in disease, the normal equilibrium of these processes is disrupted.
This paper by Cook, Wu, Odeh, and colleagues provides a nice demonstration across a variety of experimental platforms (purified protein, cell culture, in vivo) that polyGR proteins from the C9ORF72 repeat expansion mutation can accelerate aggregation of isolated, purified TDP-43, and can drive mislocalization through altering nucleocytoplasmic transport factors. This was specific to polyGR, and was not seen with polyGA, at least for purified proteins. Other DPRs were not examined.…More
Several interesting questions remain for further exploration, such as what drives TDP-43 pathology in non-C9ORF72 cases of FTD/ALS where DPRs are not present, and how this particular mechanism interacts with the many other reported toxicities of polyGR in experimental models by this group and many others. Regardless, there is a convergence of evidence from experimental systems and human pathology studies suggesting that polyGR is a key pathologic species in C9ORF72-related ALS. This study shows it could even be a more direct driver of TDP-43 pathology than previously thought.
University of Sheffield
This impressive new paper from the Petrucelli group presents a unified hypothesis linking key molecular phenotypes of C9ORF72-ALS to a single mechanism. They present evidence that poly(GR) protein translated from the C9ORF72 expansion can accelerate the aggregation of purified TDP-43. But more than that, they show that, in vivo, poly(GR) disrupts nucleocytoplasmic transport of TDP-43 and actively recruits mislocalized TDP-43 into stress granules. Thereby, the researchers have shown that poly(GR) is sufficient to reproduce all of the important aspects of TDP-43 pathology seen in human patients.
But a mechanism that is sufficient is not necessarily the key mechanism at work. Phrased another way, if we could prevent the formation of poly(GR) in human patients, would this prevent TDP-43 pathology and neuronal death? To answer this, the researchers moved to their previously published mouse model of C9ORF72-ALS (Chew et al., 2015), where they intervened to deplete sense transcribed GGGGCC-repeat RNA and sense dipeptide repeats (including poly-GR) using c9 antisense oligonucleotides, which are currently in clinical trial in ALS patients. As expected, depletion of G4C2-repeat RNA reduced levels of poly(GR) and other sense dipeptide repeats, reduced TDP-43 pathology, and reduced neuronal loss.…More
However, there are a few outstanding concerns: The Chew et al. mouse is not a perfect model of C9ORF72-ALS in that there is no reported motor neuron loss, despite molecular pathology within motor neurons. Moreover, the phenotype in these mice is comparable to FTD but not closely comparable to ALS and there is no reduction in survival. The C9ORF72 mouse that most closely models an ALS phenotype, including TDP-43 pathology and reduced survival, is from the Ranum group (Liu et al., 2016). This model is notable for increased antisense transcription of the C9ORF72 expansion in vulnerable neuronal populations. This raises the question that perhaps antisense-specific transcripts and dipeptide repeats (poly(PR), poly(PA) are key to development of C9ORF72-ALS, which obviously excludes a poly(GR)-centric mechanism.
We await the results of the current c9ASO clinical trial, but this paper gives reason to hope that the treatment will reverse TDP-43 pathology and neuronal death. In future, even more effective treatments may be devised to target poly(GR) directly.
References:
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A, Overstreet K, Edbauer D, Rademakers R, Boylan KB, Dickson DW, Fryer JD, Petrucelli L. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015 Jun 5;348(6239):1151-4. Epub 2015 May 14 PubMed.
Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron. 2016 May 4;90(3):521-34. Epub 2016 Apr 21 PubMed.
This paper by Cook et al. describes very exciting results. The authors present strong evidence that poly(GR) promotes TDP43 pathology indirectly by inhibiting its nuclear transport and more directly through physical interaction promoting TDP43 aggregation. I would be interested to know if the interaction between TDP43 and poly(GR) could be inhibited by small molecules or peptides and if so, whether this would rescue poly(GR)-mediated toxicity.
University of Massachusetts Medical Center at Worcester
This paper by Zhang et al. elegantly builds upon, and strengthens, their previous findings that progranulin is required to suppress aberrant microglial activation in the aging brain (Lui et al., 2016). For many years microglia had been overlooked as direct contributors to the pathological progression of neurodegenerative disorders, but many studies, like this one, continue to add to a growing pool of evidence that increasingly demonstrates microglia to be key drivers of neurodegeneration.
Their single nuclear RNA sequencing results demonstrate that progranulin-deficient microglia undergo a shift from a homeostatic to a disease-specific transcriptional state—akin to, yet distinct from, the disease-associated microglia (DAM) populations that have been observed in AD and ALS. Using a combination of in vitro techniques and mouse genetics, the authors further demonstrate that Grn-/- microglia promote TDP-43 proteinopathy, nuclear pore defects, and cell death in Grn-/- excitatory neurons, all of which could be mitigated by blocking complement activation.…More
These findings further elevate the complement system as a potentially viable therapeutic target for PGRN-mediated FTD, though there is still much to untangle in the relationship between the complement cascade and neurotoxicity. For example, the finding that Grn-/-;C1qa-/-;C3-/- have near-complete rescue of microgliosis may be attributable to impaired formation of the C5 convertase and its subsequent pro-inflammatory activity via C5a (An et al., 2018)—conceivably linking C1qa/C3 deletion indirectly to neuroprotection.
The authors also point out that, while their results support that blocking complement activation can mitigate Grn-/- microglia toxicity, other cell-intrinsic defects such as lysosomal dysfunction may also propagate neurodegeneration. Indeed, their data demonstrating that TDP-43 granules preferentially attach to lysosomes in Grn-/- neurons (Extended Data figure 8a), as well as their data showing that Grn-/- microglial conditioned media did not further exacerbate Grn-/- cortical neuron apoptosis in vitro (Extended data Figure 7b and 7c) each demonstrate neuron-intrinsic deficits. Additionally, TDP-43 cytoplasmic localization and nuclear pore dysfunction were each much more pronounced in vitro when both microglia and neurons were Grn-deficient.
Although progranulin haploinsufficiency is known to cause FTD, and heterozygous mice were not examined in detail, this study provides key insights into the way progranulin-mediated maintenance of microglial homeostasis may be contributing to FTD progression. Moving forward it will be important to continue to dissect the respective contributions of various CNS cell types in the pathogenesis of FTD and related degenerative disorders.
References:
Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC, Martens LH, Gao F, Coppola G, Sloan SA, Hsieh CL, Kim CC, Bigio EH, Weintraub S, Mesulam MM, Rademakers R, Mackenzie IR, Seeley WW, Karydas A, Miller BL, Borroni B, Ghidoni R, Farese RV Jr, Paz JT, Barres BA, Huang EJ. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell. 2016 May 5;165(4):921-35. Epub 2016 Apr 21 PubMed.
An XQ, Xi W, Gu CY, Huang X. Complement protein C5a enhances the β-amyloid-induced neuro-inflammatory response in microglia in Alzheimer's disease. Med Sci (Paris). 2018 Oct;34 Focus issue F1:116-120. Epub 2018 Nov 7 PubMed.
University of Pennsylvania
This study by Cook et al. nicely builds on many years of work documenting the effects of the C9ORF72 G4C2 expansion and dipeptide repeat (DPR) proteins in vivo.
Previously, it was shown that expressing the G4C2 repeat leads to RNA foci, DPRs and TDP-43 pathology. Amongst the various DPRs, this paper indicates that poly(GR) protein is likely driving TDP-43 pathology in vivo. They also found that poly(GR) and TDP-43 protein aggregates can co-localize. In human tissues, most DPR aggregates do not co-localize with TDP-43 aggregates, although we and others have documented that some DPR aggregates do co-localize with TDP-43 inclusions where the DPR forms a central core which is coated with TDP-43 protein. Importantly, antisense oligonucleotides (ASOs) were able to reduce levels of poly(GR) and TDP-43 inclusions.…More
A robust literature suggests that proteins, including RNA-binding proteins such as TDP-43, can undergo liquid phase separation, which is mediated through low complexity domains and modulated by RNA binding. Intriguingly, the authors find that TDP-43 co-aggregation with poly(GR) is not dependent on the low-complexity domain or RNA binding. This highlights that there may be diverse mechanisms that lead to TDP-43 inclusion formation.
A longstanding argument against the hypothesis that DPRs drive toxicity is human neuropathology data, which shows that some regions, such as the cerebellum, do not degenerate but exhibit abundant DPR aggregates. There is an overall poor correlation between DPR inclusion burden and neurodegeneration. I think this study suggests that there may be an interaction between DPRs and TDP-43. So perhaps DPR accumulation promotes TDP-43 inclusion formation in cell types that are predisposed to TDP-43 dysfunction. In contrast, perhaps DPRs may be relatively innocuous in regions and neurons that are resistant to TDP-43, such as the cerebellum.
This interaction between DPRs and TDP-43 could be one reason why the DPR burden in postmortem human tissues does not correlate with neurodegeneration.
Make a Comment
To make a comment you must login or register.