C9ORF72 Toxicity Tied to Mitochondria, Transcriptional Machinery
Quick Links
Two studies offer new potential ways of quelling the neuron-killing rampage of the C9ORF72 hexanucleotide expansion, the biggest genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). In one, a mouse model featuring a slow buildup of the C9ORF72-encoded poly(GR) dipeptide points to mitochondria as early casualties, well before appreciable neurodegeneration or behavioral symptoms. Led by Fen-Biao Gao, University of Massachusetts Medical School in Worcester, scientists were able to reverse early damage by turning off poly(GR), or by boosting levels of a mitochondrial protein. The work appeared May 13 in Nature Neuroscience.
- Inducible C9ORF72 mouse model of ALS/FTD shows slow, reversible phenotype.
- Harmful poly(GR) proteins poison mitochondria.
- Newly identified transcription machinery may become therapeutic target.
In the other, Nancy Bonini’s lab at the University of Pennsylvania in Philadelphia uncovered a way to nip C9ORF72 expression in the bud and prevent production of toxic RNAs and dipeptides altogether. Their paper, published May 20 in Nature Neuroscience, identifies PAF1C, a transcription complex needed to read the C9ORF72 gene expansion RNA in flies and yeast. PAF1C components were upregulated in brain samples from people with C9ORF72-FTD, and bound to the C9ORF72 promoter. The role of PAF1C in expression of the expanded repeats suggests its human homolog may be a target for mitigating C9ORF72 toxicity.
In ALS or FTD caused by the C9ORF72 expansion, multiple copies of a pathogenic DNA hexanucleotide give rise to toxic RNAs, whose translation produces dipeptide repeat proteins (DPRs) that somehow clog up cells. Poly(GR) is a particularly damaging DPR in both animal models and cells, associating with ribosomes and poisoning protein synthesis (Jun 2018 news; Jun 2018 news).
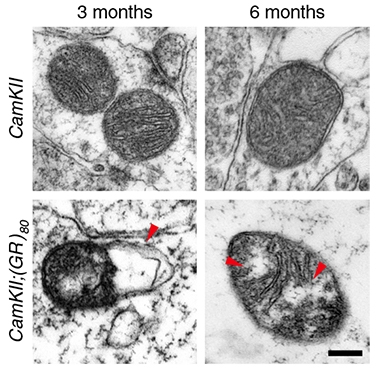
Early Breakdown. Mitochondria show signs of stress in mice with low-level poly(GR) expression. Normal mitochondria (top) are plump and filled with well-ordered inner membranes; mitochondria from poly(GR) mice are misshapen, with disrupted membranes (red arrows, bottom panels). [Courtesy of Choi et al., Nature Neuroscience, 2019.]
One limitation of current animal and cell models, Gao told Alzforum, is that they express poly(GR) at high levels. To approximate more physiological conditions, first author So Yeon Choi made transgenic mice that express an 80-repeat poly(GR) at a fraction of the level seen in adult ALS/FTD brain tissue. In these mice, poly(GR) accumulated slowly over time. At 3 months, the DPR was undetectable by immunostaining, though a more sensitive ELISA did pick up small amounts. By 6 months, poly(GR) was visible in a few neurons, mostly in the frontal cortex. It continued to accumulate in neuronal cell bodies' dendrites as the mice aged, although even by 8 months, most cells still showed no signs of protein aggregation. In 3- to 7-month-old mice, poly(GR) maxed out at 15 percent of the levels in postmortem frontal cortices from people with C9ORF72 ALS/FTD.
Even at these low levels, poly(GR) wrought damage. At 3 months, all seemed normal, but by 6 months the mice started to change. They interacted less with each other and seemed more anxious, perhaps mimicking FTD’s effects on frontal cortex circuits in people. Those changes came with signs of synaptic dysfunction and neuronal death, and also astro- and microgliosis.
The finding of neuronal loss and synaptic dysfunction even before GR is visible by immunostaining is striking, said Adrian Isaacs, University College London. “This suggests relatively low levels of likely soluble GR are sufficient to induce neurodegeneration,” he wrote to Alzforum (full comment below).
What did poly(GR) do? Gao’s lab had reported previously that, in cells derived from patients with C9ORF72-ALS/FTD, mitochondria were compromised and oxidative stress was attacking DNA; other groups also tied DNA damage to the repeats (Lopez-Gonzalez et al., 2016; Feb 2019 news). This held true in the new mice. By 3 months, mitochondria in poly(GR)-bearing neurons had structural changes, which worsened with age. By 6 months, mitochondrial ATP production was down, and DNA was showing signs of damage. The investigators spotted a low-grade accumulation of nonaggregated poly(GR) inside mitochondria.
Further work established that poly(GR) preferentially bound to the ATP synthase F1 subunit alpha (Atp5a1), and triggered its degradation. The scientists discovered lower levels of Atp5a1 in the 6-month-old poly(GR) mice’s brains. Forced overexpression of Atp5a1in primary cortical neurons from the mice decreased their mitochondrial fragmentation, restored ATP levels, and prevented cell death. Importantly, they saw evidence of mitochondrial distress in cells from people with C9ORF72 ALS/FTD, where they found a similar reduction in Atp5a1 protein.
The model suggests that mitochondrial defects could be one of the earliest, if not the earliest, signs of poly(GR) toxicity, Gao told Alzforum. “At 7 months, we don’t see a lot of neuron loss, so it appears poly(GR) compromises neuronal function long before it kills neuron,” he said.
Because the transgene has a promoter that can be repressed by feeding the mice doxycycline, the investigators were able to ask what happens once they shut off poly(GR) production. Doing so starting at 1 month of age prevented both the subsequent cellular pathology and behavior changes. What’s more, even when they shut off transgene expression at 7 months, once pathology was well underway, they still saw a benefit. Two months later, cortical poly(GR), neuron death, DNA damage, microgliosis, and astrogliosis, were down; Atp5a1 was up.
This reversal has far-reaching consequences, according to Ludo Van Den Bosch, KU Leuven, The Netherlands. “It strongly indicates that therapeutic strategies lowering the expression of toxic dipeptide-repeat proteins can reverse disease phenotypes, even after disease onset,” he wrote to Alzforum (full comment below).
Even though the model shows a specific mechanism of poly(GR) toxicity, this peptide is but one of several in C9ORF72’s arsenal. Sorting out how mitochondrial toxicity fits with the numerous effects of RNA and other dipeptides will be a challenge, said Robert Baloh, Cedars-Sinai Medical Center, New York (full comment below). Laura Ranum, University of Florida, Gainesville, also noted that poly(GR) represents just a part of the picture. “In the future, it will be interesting to test if strategies described to reverse GR toxicity improve phenotypes in mouse models that express the repeat expansion and produce the multiple types of repeat-associated proteins found in patients,” she wrote to Alzforum (full comment below).

In a Fly’s Eye. A genetic screen for modifiers of GGGGCC-repeat toxicity in Drosophila eye found a transcription complex needed for production of the troublesome RNA. [Courtesy of Goodman et al., Nature Neuroscience, 2019.]
The second paper, by first author Lindsey Goodman and others, suggests that another potential route to turning off C9ORF72-repeat toxicity may be to avert its transcription in the first place. Using a genetic screen in fruit flies, the scientists sought genes that temper the toxicity of the GGGGCC hexanucleotide repeat. It pulled out several components of the PAF1C RNA polymerase III complex, which is especially good at transcribing GC-rich DNA. Knocking down PAF1C in flies reduced repeat RNA and its toxicity. The protection depended on blocking buildup of the RNA; when the scientists supplied poly(GR) peptides from other, non-repeat RNA, PAF1C knockdown did not block toxicity, nor did it block toxicity in a TDP-43 fly model.
Several components of this polymerase were required for RNA to be produced off longer repeats, but not shorter, nontoxic ones. Those same factors were upregulated in fly, mouse, and human C9ORF72-expressing cells. One component, Leo1, was more abundant in mice bearing the C9ORF72 expanded repeat; another, PAF1, was up 50 percent in C9+ patient-derived cell lines.
Chromatin immunoprecipitation placed Leo1 on the C9ORF72 DNA in C9+ human cells. Finally, PAF1 and LEO1 were upregulated in postmortem frontal cortices from people with FTD, though not ALS, and this tracked with presence or absence of cortical pathology. In brain tissue of C9+ ALS patients, the researchers noted a strong correlation between levels of Leo1 or Paf1 and C9ORF72 transcripts; this was not the case in people with C9- FTD or in healthy controls.
Based on these results, downregulation of repeat RNA transcription represents another promising avenue for therapeutic development, Gao told Alzforum.—Pat McCaffrey
References
News Citations
- In New ALS/FTD Mouse Model, Poly(GR) Peptides Poison Ribosomes
- Dipeptide Repeats May Hobble Ribosomes in C9ORF72-FTD Patient Brain
- Studies Point to DNA Difficulties in ALS/FTD
Paper Citations
- Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, Petrucelli L, Miller BL, Almeida S, Gao FB. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron. 2016 Oct 19;92(2):383-391. Epub 2016 Oct 6 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Choi SY, Lopez-Gonzalez R, Krishnan G, Phillips HL, Li AN, Seeley WW, Yao WD, Almeida S, Gao FB. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat Neurosci. 2019 Jun;22(6):851-862. Epub 2019 May 13 PubMed.
- Goodman LD, Prudencio M, Kramer NJ, Martinez-Ramirez LF, Srinivasan AR, Lan M, Parisi MJ, Zhu Y, Chew J, Cook CN, Berson A, Gitler AD, Petrucelli L, Bonini NM. Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat Neurosci. 2019 Jun;22(6):863-874. Epub 2019 May 20 PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
Neurobiology, KU Leuven & VIB
This paper nicely shows that expression of a poly(GR) in all the layers of the cortex under the control of a CamKII promotor can induce an ALS/FTD-related phenotype. A major advantage of this study is that the poly(GR) is expressed at a relatively low level and that phenotypes induced by huge overexpression of the transgene are avoided.
Another very interesting characteristic of the system used in this study is that the expression of poly(GR) can be reduced by feeding the mice with doxycycline. Interestingly, behavioral and even the cellular phenotypes can be significantly reversed when the expression of poly-GR is lowered. This is fascinating and has far-reaching consequences. It strongly indicates that therapeutic strategies lowering the expression of toxic dipeptide-repeat proteins can reverse disease phenotypes, even after disease onset. This reversal of the phenotype can be considered as a very interesting new insight, in addition to the systematic characterization of the underlying disease mechanism, which is related to mitochondrial dysfunction.
View all comments by Ludo Van Den BoschUniversity College London
This paper describes an interesting new poly(GR)-expressing mouse model. We will need a range of different models to ultimately understand the role of each DPR and their contribution to C9ORF72-repeat pathology, so this is a welcome addition.
The most striking finding is that neuronal loss and synaptic dysfunction are identified even though GR is not detectable by immunostaining until six to eight months of age (although it can be detected earlier with ELISA). This suggests relatively low levels of likely soluble GR are sufficient to induce neurodegeneration.
The paper also links poly(GR) to an early defect in mitochondrial function, which could explain the later development of DNA damage. Many pathways have been implicated in C9ORF72-repeat-induced neurodegeneration, and mitochondrial dysfunction should now be added as a new avenue for further investigation. Consistent with the previously reported AAV poly(GR) mouse model, no TDP-43 pathology was observed, and so the link between DPRs and TDP-43 is still an outstanding question.
View all comments by Adrian IsaacsDirector of Neuromuscular Medicine, Cedars-Sinai Medical Center
This is a very nice addition to the building literature on mechanisms of toxicity from DPR expression in animal models. It brings to light a surprisingly specific mechanism, whereby poly(GR) alters mitochondrial ATP5A1, further linking mitochondrial dynamics and dysfunction to neurodegeneration and providing a potential therapeutic avenue.
Challenges going forward for the field include interpreting this mechanism of toxicity in the context of the numerous other toxicities of the different DPRs, and determining whether some are more important than others, and where and when they are happening in human disease.
Regardless, it provides further support for the idea that diminishing gain-of-function products of the C9ORF72 repeat could have a therapeutic effect, and many of these strategies are in preclinical or clinical phase testing.
View all comments by Robert BalohUniversity of Florida
This work provides an additional mouse model and research tool for the C9 ALS/FTD community. Because C9 ALS/FTD disease is a complex disease, with the expression of two mutant transcripts (sense and antisense) and six dipeptide repeat-containing proteins, which are produced by repeat-associated non-AUG (RAN) translation, this polyGR-expressing mouse model is a simplified model for studying the potential contribution of polyGR to disease pathology.
The paper provides a compelling data set showing that moderate expression of polyGR causes mitochondrial dysfunction that is linked to reductions in Atp5a1. In the future, it will be interesting to test if strategies described to reverse GR toxicity improve phenotypes in mouse models that express the repeat expansion and produce the multiple types of RAN proteins found in patients. It will also be of interest to test if Atp5a1 levels are reduced in these G4C2 mouse models and also in C9 patient tissues.
View all comments by Laura RanumMake a Comment
To make a comment you must login or register.