Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
Quick Links
New research bolsters the case that brain-derived microglia need TREM2 to essentially wall off amyloid plaques, but exactly how they do that remains up for debate. As reported in the May 18 Neuron, scientists led by Jaime Grutzendler at Yale University, New Haven, Connecticut, used confocal and super-resolution microscopy to show that TREM2-positive microglia surround and encase amyloid fibrils, protecting neurons in the process. Yet TREM2 itself appears to lend little support to phagocytosis of Aβ. The technical caliber of the work and the quality of the microscopy led researchers in the AD field to call the study “stunning.” It comes on the heels of another paper, in the April 18 Journal of Experimental Medicine, which suggests the microglia that surround plaques are brain-derived, not peripheral myeloid cells as others had suggested previously. In this second paper, researchers led by Marco Colonna and David Holtzman at Washington University, St. Louis, used parabiosis, in which the blood supply of one mouse is shared with another, to show that no cells derived from the circulation corral plaques in the brain. Some researchers saw this as definitive proof that brain-resident microglia are the sine qua non for plaque wrangling, but others were not so sure. The WashU and Yale researchers collaborated on both papers.
As reported in the Neuron paper, first authors Peng Yuan, Carlo Condello, and colleagues used confocal and super-resolution stochastic optical reconstruction microscopy, or STORM (see Jan 2009 news) to examine the interaction between microglia and plaques in exquisite detail. They compared 5xFAD and APPPS1-21 mice that were homozygous, heterozygous, or null for the TREM2 or DAP12 genes. The latter partners with TREM2 to elicit downstream signaling cascades. They also examined the brains of 10 AD patients with the R47H TREM2 mutation that increases risk for the disease, and nine AD patients who had normal TREM2 genes.
Plaque Anatomy. STORM shows that with TREM2 in short supply, plaques contain more diffuse Aβ. [Courtesy of Jaime Grutzendler and Neuron.]
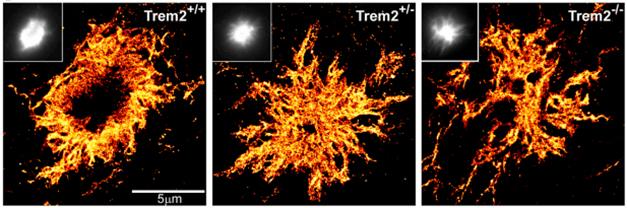
In the mice, STORM microscopy revealed a dense plaque core surrounded by a mesh-like region, all in a halo of diffuse Aβ (see image above). In animals lacking TREM2 or DAP12, the diffuse type dominated and the fibrils in these plaques branched more extensively and were thinner (see image below). These filamentous plaques came with dramatic neuritic dystrophy. Grutzendler believes that microglia encapsulate very early filamentous Aβ deposits and later more dense plaques, effectively isolating the peptide from surrounding neurons (Condello et al., 2015). “The idea, which has not been fully appreciated, is that by capping or insulating plaques, microglia change their structure to make them more inert,” he told Alzforum.

Encapsulating Plaques. In 5xFAD mice, microglia lacking TREM2 ignore amyloid plaques (cyan), which become more diffuse. [Courtesy of Jaime Grutzendler and Neuron.]
The researchers found similar structures in brain tissue from AD patients. While both R47H carriers and non-carriers had similar numbers of plaques, the carriers had twice as many filamentous ones and half as many microglial processes surrounding them. Axonal dystrophy occurred around many of the diffuse plaques, suggesting they were toxic, however they appeared extremely dim under the microscope, making Grutzendler doubt that PET imaging would even pick them up.
What role does TREM2 play in this encapsulation? In confocal imaging of brain tissue from AD patients and from the various mouse models, Yuan and Condello detected TREM2-positive processes infiltrating plaques and wrapping around amyloid fibrils. These cellular tendrils are largely absent in the R47H mutation carriers (see image below). They are also absent in the TREM2 and DAP12 knockout mice and heterozygotes, which had fewer microglia near plaques as well. The researchers concluded that TREM2 helps to polarize processes in the microglia, enabling them to reach out to contact and envelop amyloid fibrils. “The discovery of these distinct processes that contact plaques is an extremely important contribution and very elegantly demonstrated,” said Gary Landreth, who just moved from Case Western Reserve University, Cleveland, to the Stark Neurosciences Research Institute at the Indiana University School of Medicine in Indianapolis.

Contact.
Microglia (green) extend processes (arrowheads) to encapsulate plaques (magenta). This works less well in people with the R47H TREM2 mutation (right).
“This work points to loss of TREM2 function being detrimental for the brain,” said Christian Haass from the German Center for Neurodegenerative Diseases (DZNE), Munich.
That said, Haass took issue with the conclusion that TREM2 plays little role in the microglial digestion of plaques. Yuan and colleagues found that microglia in TREM2+/- mice had ingested just as much Aβ as did microglia in control 5xFAD mice, suggesting phagocytosis in the TREM2 heterozygotes is normal. Since the TREM2 variants that lead to AD are heterozygous, Grutzendler concluded that phagocytosis may be mostly normal in R47H carriers, as well. In contrast, Haass and colleagues have previously reported that TREM2 mutations compromise phagocytosis in vitro (see Alzforum webinar on Kleinberger et al., 2014). He told Alzforum he now has evidence for compromised phagocytosis by TREM2-deficient microglia in brain slices, and that loss of TREM2 compromises immunotherapy responses as well. Haass noted that the R47H mutation has the weakest effect on phagocytosis of all known TREM2 mutations tested, hinting that more profound effects might be seen in carriers of other mutations, such as those linked to frontotemporal dementia.
“It is clear from [Grutzendler’s] work that TREM2 has dramatic effects on the morphology of plaques, but the reason is open to interpretation,” Haass said. Landreth agreed, emphasizing that the authors base their conclusions about phagocytosis on static images of internalized amyloid. “Cells can clear that out in seconds,” he said. Landreth noted that the AD brain is chock full of plaque-associated myeloid cells that are inflammatory and inefficient at phagocytosis.
Grutzendler acknowledged that TREM2 mutations may affect phagocytosis since they found that complete loss of the protein reduced the amount of Aβ in the microglia. “This is consistent with data from the Haass lab showing cultured cells lacking TREM2 or expressing only mutant forms have reduced capacity to internalize Aβ,” he said. He emphasized that loss of one copy of the gene was insufficient to change plaque density, but had a marked effect on plaque structure. “While our static assay may have missed some effect on phagocytosis, our results suggests that in the case of Aβ, TREM2 may be more important for encapsulation and for making plaques inert.” Haass wondered about that. His group has correlated TREM2 signaling with biomarkers of AD and neurodegeneration in people. “We find no relationship with Aβ in the CSF, but we do find a strong correlation between TREM2 processing and total tau and phospho-tau. This suggests to us that cell death stimulates TREM2 activity.”
What about the origin of these TREM2 microglia? Previously researchers led by Landreth and Bruce Lamb at the Cleveland Clinic reported that TREM2-positive microglia express the peripheral myeloid cell marker CD45 and lack the brain microglia P2RY12, suggesting the cells around plaques came from the circulation. Others cautioned about using these markers to claim provenance since cells can turn them on and off as they respond to their environment. Colonna and colleagues set out to test the brain for cellular intruders using parabiosis, which many consider a litmus test for infiltration of peripheral monocytes to the brain.
To distinguish brain and circulating monocytes, joint first authors Yaming Wang, Tyler Ulland, and Jason Ulrich took advantage of allelic variation in the mouse CD45 gene. They connected the circulatory system of CD45.1 wild-type mice to that of CD45.2-expressing 5xFAD animals, and then looked for CD45.1 expression in the brains of the latter. They conjoined mice at four and eight months old, and tested for CD45.1 in the brain four weeks later. Wang found hardly any exchange of monocytes between blood and brain. In the 5xFAD animals, microglia surrounding plaques were predominantly CD45.2, i.e., endogenous brain cells. Similarly, they found no exchange between wild-type and APPPS1-21 mice.
“I think this is definitive proof that peripheral cells play no role in plaque clearance,” said Haass. “This should end that debate.” Landreth was not so sure, noting that the findings were negative. “Over four weeks they find no infiltration from the circulation. While there’s no reason to question that outcome, that doesn’t mean it can’t happen,” he said. Landreth thinks a definite test would require genetic models that incorporate lineage tracers to determine cell type.—Tom Fagan
References
News Citations
Research Models Citations
Webinar Citations
Paper Citations
- Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 2015 Jan 29;6:6176. PubMed.
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016 May 18;90(4):724-39. PubMed.
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016 May 2;213(5):667-75. Epub 2016 Apr 18 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.