Pittsburgh Compound-B Zooms into View
Quick Links
For several years now, the Alzheimer's research and treatment communities have been awaiting the fruition of promising research into quantitative imaging agents that could signal the presence of amyloid in the brains of living people (see ARF meeting reports from Stockholm and Paris). Such a biomarker could become a diagnostic tool, test the amyloid hypothesis definitively in live humans, and help assess whether experimental treatments work in trial populations. Now one of the hot prospects—the thioflavin derivative termed Pittsburgh Compound-B (PIB)—has shown its ability to distinguish clinically diagnosed patients from control subjects.
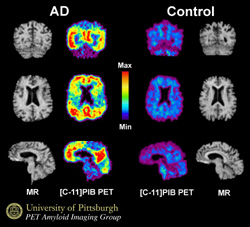
Bill Klunk and Chester Mathis, who developed the hydroxylated benzothiozole as a PET tracer together at the University of Pittsburgh in Pennsylvania, published the study today in an early online article in the Annals of Neurology. Their collaborators Henry Engler, Bengt Langstrom and Agneta Nordberg at Uppsala University in Sweden, and others, evaluated PET images from 16 patients diagnosed with mild AD and nine controls (six older subjects, and three young subjects who almost certainly had no amyloid deposits). In the AD patients, the PIB signal doubled relative to controls (meaning they retained significant amounts of PIB) in various cortical areas, particularly in frontal cortex, as well in the striatum. By contrast, the biomarker sailed through the brains of the control subjects, clearing out without accumulation. Areas not affected by amyloid deposition in AD (subcortical white matter, pons, and cerebellum) did not differ between the groups.
View larger version (Close the window to return to news story.)
Are you looking at the future of Alzheimer's diagnosis? In these PET images from Bill Klunk, Chester Mathis, and colleagues, their tracer Pittsburgh Compound-B (PIB) lights up cortical areas laden with amyloid in AD patients. The results of a PIB-PET study conducted at Uppsala University in Sweden appeared today in the online edition of the Annals of Neurology. As it happens, the work of Klunk, Mathis, and their collaborators was featured tonight in the PBS television special called "The Forgetting: A Portrait of Alzheimer's."
Interestingly, the only control subject to show PIB signals in the same range as the AD patients was the oldest (77 years), leading the authors to ask whether this person might be in a preclinical stage of AD. As the authors point out, "The ability to longitudinally follow PIB retention as an in-vivo measure of amyloid deposition now gives us a tool through which we may be able to answer this question in a manner that postmortem studies can not." Conversely, three AD patients had PIB values in the range of the control group. Their clinical deficit was mild, and they did not progress significantly over the two- to four-year follow-up period.
The researchers compared their PET results to several other measures. For example, they found an inverse relationship between PIB signal and glucose turnover in parietal cortex in the AD patients, but no correlation between PIB signal and MMSE scores or ApoE4 genotype. This was a small, proof-of-principle study.
The authors close their article with a warning against the circular reasoning that is inherent in the acceptance of amyloid deposition as both a putative cause and diagnostic proof of AD. At this early stage of imaging research, they prefer to think of their method as a way to investigate β-amyloidosis in the brain. With this precept, they write, "Several basic, unbiased questions then can be asked regarding (1) the correlation of β-amyloidosis with clinical diagnosis; (2) the natural history of β-amyloidosis and its onset relative to clinical symptoms of dementia; and (3) the ability of β-amyloidosis to serve as a surrogate marker of efficacy for anti-amyloid therapeutics.
A second human imaging study with PIB is ongoing at the University of Pittsburgh. At the 33rd Annual Meeting of the Society for Neuroscience held last November in New Orleans, Klunk reported some initial data of this study. It builds on the Swedish study by including people with mild cognitive impairment (MCI), a prodromal stage of AD, and by analyzing PIB pharmacokinetics in blood samples drawn from the study participants. As in the Swedish study, AD patients, but not controls, retained significantly more PIB in brain areas known to accumulate amyloid, Klunk said. MCI subjects fell in between and varied in their PIB retention; early results of direct comparisons with MRI imaging hint that PIB-PET is more sensitive at picking up MCI than is MRI. The scientists will follow the Pittsburgh subjects over time to see how their PIB signal and their clinical status changes. One hope of such longitudinal studies is that the time of diagnosis could be pushed back years into the preclinical phase. Klunk noted that even if all future studies go well, a more practical radioligand must first be developed before PIB can become widely available.
Klunk told the audience that the University of Pittsburgh has signed a licensing agreement with Amersham Biosciences to move PIB through clinical development, but added that this agreement does not restrict academic collaborations. Several centers in the U.S. have agreed to test PIB for research purposes.
And now, for the perennial optimists: At the conference, other scientists not connected with this work privately mused that together, this and other developments in experimental therapeutics conjure up the vision of a patient complaining about subtle memory problems and visiting a neurologist. If a PIB-PET scan revealed significant amyloid buildup, the patient could receive a vaccination in the hospital to clear it out in an intense one-time treatment not unlike chemotherapy or heart angioplasty. After that, the person would be titrated to the proper dose of a statin or an NSAID, with a generous helping of antioxidants thrown in, and monitored periodically to ensure the amyloid does not come back. Science fiction? For now, yes.—Hakon Heimer and Gabrielle Strobel
References
News Citations
- Stockholm: Visualizing Amyloid Biggest Draw at Imaging Symposium, Consensus Sought on Validation
- Bill Klunk Reports from Paris on The Living Brain and Alzheimer’s Disease
Other Citations
Further Reading
News
- Amyloid Ligand Looks Suited for Future Diagnostic Test
- PET Diagnosis Poised for Prime Time? FDA Wants Consensus, Better Trials
- Bill Klunk Reports from Paris on The Living Brain and Alzheimer’s Disease
- Seeing Alzheimer’s: Advances in Brain Mapping Bring Goal a Step Closer
- PET Reduces Alzheimer Misdiagnosis
- Stockholm: Visualizing Amyloid Biggest Draw at Imaging Symposium, Consensus Sought on Validation
- New PET Probe to Aid Diagnosis and Monitoring of Alzheimer's Disease
- PET Found to Be a Specific and Sensitive Tool for Diagnosis of Alzheimer's Disease
Primary Papers
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004 Mar;55(3):306-19. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
�
PIB-PET probing is a very significant step foreward on the road to early Alzheimer diagnosis. The authors deserve sincere congratulations on this significant contribution. However, in order to be generally applicable new techniques should be affordable, which in case of PET scan is not (yet?) the case.
Moreover, we must perhaps focus most of all on the soluble Abeta mayloid fraction to target the main culprit in its early phase, before structural synaptic disturbance, and even before GSK-3 or CDK-5- mediated induction of neurofibrillary tangle accumulation, which then disrupt neurons. More effort is needed in the field of early biomarkers both of Abeta and specific hyperphosphorylated tau. These should be corallated with the authors PIB-PET or (soon to come ?) PIB-II-MRI findings.
Columbia University
The ability to visualize disease has long motivated and driven the history of Western medicine. The end of the nineteenth century represents a turning point in the ability to do so: At around the same time neuroanatomists perfected staining techniques that made disease visible under the microscope, Wilhelm Roentgen introduced the x-ray, which allowed internal structures to be seen in living patients. In 1906, a few years after Roentgen received the first Noble prize in physics, Alois Alzheimer described amyloid plaques and neurofibrillary tangles—the histological features of his eponymous disease. Now, almost a century later, these two technical developments—in-vivo imaging and in-vitro features of Alzheimer’s disease (AD)—have finally converged. In a landmark study published in this month’s issue of the Annals of Neurology, William Klunk and his colleagues show that amyloid plaques can be visualized in the living brains of AD patients.
In the reported study, they used a radio-labeled hydroxybenzothiazole, termed PIB (Pittsburgh compound B), which selectively binds to aggregated fibrillar Aβ deposits. PIB was intravenously injected into AD patients and healthy controls, and positron emission tomography (PET) was then used to image PIB retention from different gross anatomical regions. As a group, AD patients were observed to have greater PIB retention measured from the frontal, parietal, and temporal cortices, with no difference observed in the cerebellum. The greatest difference between the groups was observed in the frontal cortex, while within the temporal cortex, a greater difference was observed in the lateral temporal lobe compared to the medial temporal lobe. The authors further demonstrated that PIB retention correlated with regional basal metabolism, as detected with PET measures of glucose uptake.
Taken together with a prior study by Shoghi-Jadid et al., Klunk’s group has unquestionably achieved the century-old goal of visualizing amyloid plaques in living subjects. With this conquest, we can begin to assess the ultimate utility of this approach. A range of imaging techniques have been developed attempting to enable researchers to visualize different features of AD—volumetric changes measured with MRI; metabolic changes measured with PET, SPECT, and fMRI; and now, histological changes measured with PET. In general, in-vivo imaging is needed to address three outstanding clinical questions:
1. How do we improve our ability to dissociate mild forgetfulness caused by early, pre-dementia AD from mild forgetfulness caused by normal aging? This is a question of early detection.
2. How do we improve our ability to dissociate dementia caused by AD from other dementing illnesses? This is a question of diagnostic specificity.
3. What is the best way to test for drug efficacy? This question is important both for drug development as well as for following the course of approved drugs.
In this regard, the Annals paper demonstrates that the precision and integrity with which the Klunk group perform their groundbreaking science extends also to their scientific reporting. For example, they highlight the fact that, although group differences were detected, there was significant overlap between AD and controls, suggesting that, at this point, imaging plaques might not be appropriate for early detection and early diagnostics. Nevertheless, although not explicitly assessed in their study, it seems plausible that imaging amyloid plaques will aid in enhancing diagnostic specificity when presented with an atypical demented patient. Furthermore, imaging amyloid plaques should aid in testing drugs designed as "plaque busters." The authors highlight an additional utility of their approach: By being able to quantify brain β amyloidosis (a term they borrow from George Glenner’s original studies), they can begin to cross-correlate plaque load against various factors—aging, disease onset, risk factors, etc. In so doing, they can ensure that this imaging approach will likely contribute to understanding basic mechanisms of plaque formation.
References:
Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002 Jan-Feb;10(1):24-35. PubMed.
Comment by Jorge R. Barrio, Gary W. Small, Henry Huang, and Michael E. Phelps
The pathological aggregation of the β amyloid peptide into fibrillary senile plaques (SPs) and the hyperphosphorylation of the tau protein into neurofibrillary tangles (NFTs) play a central role in the pathogenesis of Alzheimer’s disease (AD). The extent and the pattern of distribution of both lesions are indicators for the progression of AD. The initial neuropathological processes—particularly the formation of NFTs—occur in the medial temporal lobe, expanding later to the rest of the temporal lobe, the parietal lobe, and finally engulfing the whole neocortex in the late stages of disease. It is the prospect of in-vivo visualization of these neuropathological lesions that has driven the Pittsburgh group (e.g., Klunk et al., 1994), the UCLA group (e.g., Shoghi-Jadid et al., 2002), the U. Penn group (e.g., Kung et al., 2003), and other investigators to search for imaging biomarkers of these pathologies. The ideal AD imaging biomarker should be specific for the intended molecular targets (e.g., amyloid and/or tau aggregates), clear well from nonspecific binding areas (i.e., have low general lipid binding, like white matter), and yield a good signal-to-noise ratio for amyloid/tau to nonspecific sites. All this assumes that the probe binds to the aggregate site(s) in a saturable and specific manner, similar to neuroreceptor binding, although it is now apparent that amyloid and tau aggregates are complex conglomerates that contain multiple binding sites with different affinities for probes (e.g., [F-18]FDDNP binds at sites different from thioflavin probes in general). Several questions come to mind in this endeavor. First, can in-vivo imaging procedures with PET permit quantification of regional amyloid (or tau) aggregate concentrations throughout the brain? And second, can effective tracer kinetic models be established and validated to delineate transport of the labeled probe between plasma and tissue, as well as nonspecific and specific binding of the probe to amyloid and/or tau in the brain?
The early success with the use of [F-18]FDDNP (Shoghi-Jadid et al., 2002) to visualize NFTs and SPs in AD, and this work by Klunk at al. (2004) on amyloid labeling, offer an unprecedented opportunity to follow the neuropathological evolution of AD in living subjects. We should all bear in mind, however, the important challenges ahead for all amyloid/tau probes under development. The opportunity they provide is not only in early diagnosis, but also in early and repeated monitoring of both amyloid and tau anti-aggregation therapies with newly developed drugs—an active area of research and development in the pharmaceutical industry. In-vivo visualization of these brain pathologies will also help develop further understanding of how anti-aggregation drugs—like the unsuspected NSAIDs or new ones—directly interact with neurofibril aggregates (Agdeppa et al., 2003).
The article by Klunk et al. (2004) reports clinical results of the Pittsburgh Compound-B (PIB) labeled with C-11 (half-life = 20 min.) for a group of AD and control subjects. Results are encouraging; however, the authors note several methodological issues that closely relate to some of the aspects discussed above. For example, brain accumulation of PIB in AD subjects and controls is reported as SUV (standardized uptake value: tracer uptake in tissue normalized by bodyweight and injected activity) at 40-60 minutes after injection of the tracer. This approach has inherent drawbacks because it is subject to differences in fat content, bone mass, and peripheral metabolism among individuals, all of which are variable in elderly patients. Thus, apparent brain accumulation can be skewed by these factors without an independent means to verify the magnitude of these effects. Significant variability in the data can be expected because of this approach.
The authors acknowledge that they resorted to the use of SUV due to the inapplicability of the Logan graphical method (with the cerebellum as the reference region). The Logan method is only applicable when dynamic equilibrium of the tracer is reasonably achieved. The authors point out that equilibrium is not achieved at 60 minutes after injection of PIB and, in the absence of equilibrium, interpretation of SUV measures may be arbitrary. PIB equilibrium would be likely at later times, beyond 60 minutes, but the data has not been presented as of yet. An inherent limitation for longer scanning times is the short half-life of C-11, the radiolabel for PIB. Therefore, full characterization of the in-vivo kinetics of the probe remains somewhat challenging.
In light of the above issues, interpretation of the resulting data in AD patients is difficult. One of the issues is the frontal accumulation of PIB observed in some AD patients. In the discussion section, it is stated, "Antibodies to Aβ, or thioflavin S, do identify frontal cortex as a brain area very high in amyloid deposition…" as one possible explanation for the PET-PIB signal in frontal cortex in AD patients. However, the cited neuropathological studies do not confirm the importance of frontal Aβ deposition in AD. One of those references notes that, "Temporal and occipital lobes had the highest amyloid plaque densities, limbic and frontal lobes had the lowest, and parietal lobe was intermediate." (Arnold et al., 1991). Indeed, the authors recognize in the discussion section that the frontal lobe accumulation of PIB could be an artifact. The intense accumulation of PIB in white matter areas (1.5 times higher than cerebral cortical grey matter in normal subjects) indicates the high nonspecific lipid binding of PIB and would certainly be a factor to consider in measuring cortical amyloid-specific binding of PIB. This is particularly important in early stages of AD, with presumably lower amyloid concentrations, but this is not discussed in this publication. Partial volume effects (e.g., spillover of activity from one area to the other) could be very significant in this case because of cortical atrophy present in aging and AD patients. Partial volume effects are important to consider with all imaging probes when examining brain cortex in AD, particularly at later stages of disease, but can be more of an issue with relatively high concentrations of PIB in neighboring white matter. The reported net accumulation of PIB in the cerebellum, an area known not to have amyloid plaque deposition in early AD, further indicates nonspecific (or other target) binding of PIB. In parallel, it is interesting that no correlations between cortical PIB binding with MMSE scores or ApoE status had been established with AD subjects in this work.
AD pathology offers a new and unique environment for imaging with PET with its own set of unique challenges, some of which were discussed above. The authors should be commended for their efforts and congratulated for their successes. We are well aware of the difficulties and the magnitude of the task at hand. We should all be greatly encouraged by the significant progress made on amyloid imaging in humans in the last few years. This work adds to that. It reflects the great opportunities ahead for the use of molecular imaging techniques to aid in the early differential diagnosis of the various forms of dementia, and to help guide the development of therapeutic interventions by providing direct biological assessments of the brain in living patients throughout the course of disease.—Jorge R. Barrio, Professor of Molecular and Medical Pharmacology; Gary W. Small, Professor of Psychiatry; Henry Huang, Professor of Molecular and Medical Pharmacology, Professor of Biomathemathics; and Michael E. Phelps, Professor and Chairman, Molecular and Medical Pharmacology, UCLA School of Medicine.
References:
Klunk WE, Debnath ML, Pettegrew JW. Development of small molecule probes for the beta-amyloid protein of Alzheimer's disease. Neurobiol Aging. 1994 Nov-Dec;15(6):691-8. PubMed.
Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002 Jan-Feb;10(1):24-35. PubMed.
Kung HF, Kung MP, Zhuang ZP, Hou C, Lee CW, Plössl K, Zhuang B, Skovronsky DM, Lee VM, Trojanowski JQ. Iodinated tracers for imaging amyloid plaques in the brain. Mol Imaging Biol. 2003 Nov-Dec;5(6):418-26. PubMed.
Agdeppa ED, Kepe V, Petri A, Satyamurthy N, Liu J, Huang SC, Small GW, Cole GM, Barrio JR. In vitro detection of (S)-naproxen and ibuprofen binding to plaques in the Alzheimer's brain using the positron emission tomography molecular imaging probe 2-(1-[6-[(2-[(18)F]fluoroethyl)(methyl)amino]-2-naphthyl]ethylidene)malononitrile. Neuroscience. 2003;117(3):723-30. PubMed.
Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex. 1991 Jan-Feb;1(1):103-16. PubMed.
University of Pittsburgh
Response by Bill Klunk, Chet Mathis, and Julie Price
We would like to thank Drs. Otte, Scott Small, and the UCLA group for their thoughtful comments on our recent paper. We acknowledge Dr. Otte’s point that the expense of PET precludes its use as a population screening tool and more work is required in that area. The value of this technology will ultimately be weighed against other economic forces in determining its breadth of applicability. The increasing use of FDG-PET in the diagnosis and follow-up of cancer suggests economic value, but this may only be realized in Alzheimer’s disease if the imaging is tied directly to the use of effective therapies. Soluble Aβ does appear to be a valid target as Dr. Otte suggests, but we must keep in mind that soluble, oligomeric Aβ exists in equilibrium with monomeric and fibrillar Aβ. Insoluble Aβ constitutes over 99 percent of the Aβ present in AD brain; it will likely prove impossible to decrease the level of soluble Aβ over the long term without first decreasing the amount of insoluble Aβ.
Dr. Scott Small eloquently puts our work into historical perspective and into perspective with current neuroimaging technologies. Implicit in his remarks, and worthy of further emphasis, is the fact that amyloid-imaging is a technique that is complementary to existing structural (MRI) and functional (FDG-PET, blood flow, fMRI) imaging techniques. No one imaging technique will serve all purposes, but will need to be used in conjunction to answer the important questions of early diagnosis and diagnostic specificity, as well as for assessing and following the effects of new drugs. For example, while it may be obvious why one would use amyloid-imaging to assess the effectiveness of an anti-amyloid therapy such as a β-secretase inhibitor, it may make little sense to use amyloid-imaging to evaluate a more general neuroprotective drug. We appreciate Dr. Small's emphasis of our point that amyloid-imaging is first a measure of β-amyloidosis. As he suggests, the job of relating β-amyloidosis to the early diagnosis and natural history of AD remains to be accomplished.
We appreciate the congratulations and encouragement of the UCLA PET group. While there are many difficulties involved in the development of amyloid-imaging radiotracers, we share the common goals of improving early diagnosis and facilitating drug development for the benefit of those who suffer from AD and those who care for them. We regard their comments as a constructive challenge to further understand the strengths and limitations of all amyloid-imaging technologies. Towards this goal, we are in the process of performing new studies at the University of Pittsburgh and elsewhere to extend the studies presented in the Annals of Neurology paper. We are specifically addressing the methodological issues raised, and are in the process of identifying and validating a simple pharmacokinetic method for routine assessment of amyloid deposition while expanding our human studies. In the end, the field in general will decide these issues as the technologies disseminate beyond their point of origin.
I thank the authors for not going overboard with this paper. Their conclusion (from the abstract) is reasonable: "The results suggest that PET imaging with the novel tracer, PIB, can provide quantitative information on amyloid deposits in living subjects."
Fair enough.
Then, to the caveats. It is no secret that the human brain may be burdened with a huge plaque load, seen by autopsy, in the absence of cognitive deficits prior to death. PIB-PET may just as well come to prove the irrelevance of amyloid burden.
In Finland, to my knowledge, there are two PET scanners, both located in Turku. Even if we sent 100,000 people with memory impairment to Turku, the two scanners would not be enough to scan them all, let alone the baby boomers who will soon start to reach the age where they start to develop dementia.
View all comments by Mikko LaaksoMake a Comment
To make a comment you must login or register.