If the setback of the Expedition 3 study of solanezumab set their mood in the beginning, attendees at the 2016 Clinical Trials on Alzheimer’s Disease conference soon bucked up and voiced renewed determination to tackle this illness. Many took positive secondary outcomes and trends in this Phase 3 trial as a sign that this or more potent anti-amyloid drugs given earlier will make a bigger dent in the disease process. Short of major surprises, the meeting reaffirmed the sense that AD will only be slowed by deploying potent drugs at the right target (probably more than one), with the right dose, and certainly at the right time.
CTAD: Solanezumab Seen to Nudge AD Ever so Slightly
Presenting the most eagerly awaited data at this year’s Clinical Trials on Alzheimer’s Disease meeting (CTAD) held December 8-10 in San Diego, Lawrence Honig, Columbia University, New York, reported that the Expedition 3 Phase 3 trial of Eli Lilly’s solanezumab was not a total bust. The company had given up on development of the therapy for mild AD last month following release of the topline result that treatment failed to slow cognitive decline (see Nov 2016 news). Still, researchers want a breakdown of the data to see if they sustain hope for ongoing prodromal and prevention trials that are further testing this therapeutic Aβ antibody.
After intense discussion, many CTADeers took solace in positive secondary outcomes and a trend on the primary outcome. Others consider this data to be a bellwether of the amyloid hypothesis. “I do not see this as a refutation of the amyloid hypothesis, but a confirmation of it,” said Paul Aisen, Alzheimer’s Research Therapy Institute at the University of Southern California, San Diego. Aisen co-organizes CTAD and is a principle investigator on the A4 secondary prevention trial that is still enrolling to test solanezumab in asymptomatic but biomarker-positive participants. He took heart in similar trends seen across all Expedition trials. “All three show separation of curves—a consistent story—showing that treatment slows decline by a small amount,” he noted during a panel discussion. Others were less convinced. “Results from these three negative solanezumab trials neither prove nor disprove the amyloid hypothesis,” said Lon Schneider, University of Southern California, Los Angeles.
Expedition 3 recruited 2,100 people with mild AD at 210 sites in 11 countries. Volunteers had to test positive for brain amyloid by either CSF analysis or a florbetapir PET scan. Half received intravenous infusions of 400 mg solanezumab every four weeks for 19 months, the other half got mock infusions. Considering how invasive and burdensome this procedure is, Honig was impressed that about 85 percent of participants in both arms completed the trial.
How, then, did the data break down? For the primary outcome, the ADAS-Cog14, patients on solanezumab declined 11 percent less than did those on placebo (p= 0.095). The curves showed a statistically significant separation between the active and placebo arms at week 28, and this continued at weeks 40, 52, and 64, but not at week 80.
Secondary outcome measures showed similar trends, and a few were statistically significant at the end of the trial. On the MMSE by week 80, people on solanezumab had declined 13 percent less than those on placebo (p=0.014). On the ADCS-iADL, the active group did better starting at week 64 (p value by week 80=0.019). On the CDR-SB, they declined 15 percent less by week 80 (p=0.004). The separation was weaker on the Functional Activities Questionnaire (FAQ). Aisen said this was no surprise because this is a relatively new scale; still, the trend favored the treatment arm at weeks 52 and 80. “In summary, the primary and all secondary [clinical] outcomes directionally favored solanezumab, though the magnitudes of the differences were small,” said Honig. He did not present data on seven other secondary markers because analysis is ongoing, he said.
Despite their disappointment at the overall result, scientists at CTAD lauded Lilly for persevering with the therapeutic strategy. Rachelle Doody, who recently moved from Baylor College of Medicine, Houston, to Roche/Genentech, thanked Eric Siemers and his colleagues at Eli Lilly for helping the field understand how to run clinical trials and for showing that amyloid is a valid treatment target. Based on the pooled analysis of patients with mild AD in the Expedition 1 and 2 trials, many had expected a positive result. Lilly powered Expedition 3 to detect a significant slowing of cognitive decline by doubling the number of patients with mild AD in Expedition/Expedition 2 and by using amyloid scans to weed out the 30 percent or so of people who had been clinically diagnosed with mild AD but turned out to have something else.
So what went wrong? In the end, the drug was simply too weak at that dose, experts agreed, and that showed up in the statistics. “Expedition 3 missed [its endpoint] because when the effect size is small, statistical significance typically varies from one trial to another,” said Aisen. Some wondered if the choice of primary endpoint was wrong, given that the CDR-SB showed a stronger effect. (This, incidentally, was also true in the negative LipiDiDiet trial presented later by Tobias Hartmann, Saarland University, Homburg, Germany.)
Honig rejected that idea. “Cognition sometimes looks better than function and other times it’s the reverse,” he said. “What we really want is clinical meaningfulness.” Aisen agreed. “In mild AD, the ADAS-Cog and the CDR-SB have similar power to detect treatment effects, and either may seem better than the other. It really depends on effect size,” he said. “The current standard is to rely on cognition first,” he added. Interestingly, Siemers noted that the placebo group declined faster than expected, which may reflect the Aβ-selection criteria and more active disease, he said, but he did not address whether that affected the outcome of the trial.
Others, noting that the trial came oh-so-close, wondered if the dose was right. Siemers agreed a higher dose may have been effective, but countered that safety was a major factor in settling on 400 mg. When asked if Lilly would have filed for FDA approval had the primary outcome been positive, Siemers said that would have been a major discussion. When Lilly scientists presented data on the persistence of the Expedition 1/2 small benefit on 3½-year follow-up to FDA statisticians at the 2015 AAIC conference, they received a rather pointed reminder to instead aim for a large effect size (see Aug 2015 conference news). With a small effect size, they would have had to address the question of clinical meaningfulness, Siemers said.
All things considered, some researchers at the meeting thought the outcome a blessing in disguise, since approval of a first drug, even if weak, can douse enthusiasm for developing other drugs that might turn out to be much stronger. Moreover, an expensive biologic drug of questionable effect size can attract unwelcome controversy about drug pricing and cost versus benefit.
Where does this leave the ongoing trials of solanezumab in prodromal AD? Nick Fox, University College London, England, suggested the dose be raised given that the antibody appears safe—there were no serious adverse events in Expedition 3. Only a few minor ones reached significance, such as nasal congestion and vitamin D deficiency, and some of those affected the placebo group more than the active group. Siemers said there is a lot of discussion around raising the dose but that it is not straightforward and no decision has yet been made.
Aisen said he expects the treatment effect to be larger at earlier stages of the disease. If true, this would bode well for the DIAN, A4, and ExpeditionPro trials. CTAD co-organizer Bruno Vellas, University Hospital of Toulouse, France, agreed that earlier will be better, but emphasized the medical need of the millions of people currently living with mild AD. “We have to continue to develop treatments for mild AD for the sake of those patients,” he insisted.
Some took a more cautionary stance. Steve Salloway, Brown University, Providence, Rhode Island, warned against putting much faith into the secondary outcome measures since the statistics were not corrected for multiple comparisons. Honig argued that correction was not required, since Lilly was not trying to claim efficacy based on those secondary measures. Suzanne Hendrix, president and CEO of Pentara Corporation, Salt Lake City, split the difference. “When the primary endpoint is not significant, one should interpret any secondary with caution,” she said, “but at some point the level of evidence of the secondary is good enough that you don’t have to question if it is real.” The secondaries in Expedition 3 were all correlated because they all measure the same underlying disease process, hence one doesn’t have to correct as much, she said. “All the evidence together says we have changed the disease process [in Expedition 3], but by only a small amount,” Hendrix concluded.
Others did not buy that the disease process was altered, because the biomarker data was weak. While a 500- to 800-fold increase in plasma Aβ40 and Aβ42 in the active group indicated the antibody bound and held the peptide in the blood, the brain biomarker data were murkier. Brain volume shrank with both drug and placebo at about the same rate, and brain ventricular volumes expanded by the same amount. Florbetapir PET at baseline and week 80 hinted that solanezumab lowered the cortical SUVR a tad, but the difference was not significant. Honig showed no CSF Aβ data because its analysis is ongoing.
The tau data troubled people the most. Both CSF total tau and p-tau rose more in the treatment than placebo group, with the former just missing significance (p=.06). Tau PET using AV1451 also hinted at higher levels in the treatment group, though again this was not significant. “The tau data is definitely concerning,” said Reisa Sperling, Brigham and Women’s Hospital, Boston, co-principle investigator on the A4 trial with Aisen. “It is impossible to know what that means in this small substudy, but I would like to see more data in the subgroup who started with lower levels of pathology,” she told Alzforum. Anton Porsteinsson, University of Rochester, New York, agreed. “That there’s no difference in density of fibrillar Aβ, and that the trend for tau is in the wrong direction is more worrying than the clinical effect size,” he said.
Honig cautioned that the data are difficult to interpret. “A drug might work and not affect plaques, or it might affect plaques and not work,” he said. “The best scenario would be if the biomarkers and clinical outcomes move in the same direction, but we don’t have complete data and because the effect sizes are small, it is too hard to judge,” he said. Aisen stressed that this antibody was not designed to attack plaques, but to target soluble Aβ. “For this antibody, the best marker may be plasma Aβ, because it shows you have tied up the soluble peptide,” he said.
Observers agreed that this trial does not reduce the likelihood that other anti-Aβ immunotherapies will succeed. “Solanezumab has no bearing on aducanumab. Different studies, different targets,” said Schneider. “I’d expect any of the anti-Aβ antibodies might work better at a preclinical stage when neurodegeneration is less extensive,” Aisen concluded.—Tom Fagan
Tau Inhibitor Fails Again—Subgroup Analysis Irks Clinicians at CTAD
The first oral communication session at this year’s Clinical Trials on Alzheimer’s Disease meeting (CTAD), held December 8-10 in San Diego, opened on a surreal note. Lon Schneider, University of Southern California, Los Angeles, first told the audience that LMTM, a reduced form of methylene blue reported to prevent aggregation of the neurofibrillary tangle protein tau, failed to slow cognitive decline in a Phase 3 trial of mild Alzheimer’s disease. This results come as no surprise, since a separate Phase 3 trial in mild to moderate AD earlier this year had also failed.
Next, however, the data were parsed in a way that left many scientists at CTAD dismayed and incredulous. Trying to provide the view of the sponsoring company, TauRx, Schneider presented a “cohort analysis,” i.e., an analysis not based on the randomized data. It showed an apparent effect of both the active and placebo treatment—which in fact contains a very low dose of the drug—in patients who were not taking acetylcholinesterase inhibitors (AChEIs), a current standard therapy for AD. In a bizarre turn, TauRx plans to abandon the higher doses and develop the placebo dose.
All experts at CTAD who saw the presentation and spoke with Alzforum said emphatically that any conclusion about monotherapy efficacy was at best based on a wrong comparison and, at worst, bogus.
CTAD co-organizer Paul Aisen, from the Alzheimer’s Therapeutic Research Institute, San Diego, told Alzforum that it was unfortunate that presentations of this and the prior Phase 3 trial suggested that there was an indication of efficacy. “This second LMTM study is consistent with the first—both studies are entirely negative,” he said. “The reported subgroup analyses are, in fact, uninformative, because they lacked an appropriate placebo group,” he added. Speaking later to Alzforum, Schneider also emphasized that the trial “did not work, period,” just as was reported last July for the first AD Phase 3 study, and again in September for a Phase 3 frontotemporal dementia trial (see Jul 2016 conference news; Sep 2016 conference news).
This second AD trial enrolled 800 patients with mild AD (MMSE 20-26) at 98 sites in 12 countries, including the United States, Canada, Australia, and in Europe. Schneider showed that half the patients, who were randomized to 100 mg LMTM twice a day for 18 months, declined on cognition (ADAS-Cog) and functional scales (ADCS ADL) just as quickly as did patients on placebo. As in the first AD Phase 3 trial, which had tested 75 mg and 150 mg LMTM twice daily, the curves for the treatment group were almost indistinguishable from those taking the 4 mg LMTM placebo. Unusual as it sounds, the Phase 3 trials admixed a small dose of LMTM, which TauRx originally claimed to be inactive, to the placebo in order to keep the study blinded. This was necessary because the drug colors the urine and would otherwise alert patients that they were in the active arm. Anecdotally, clinicians at CTAD told Alzforum that this does not work in practice, because of an obvious difference in urine color between those on the high and low doses.
Researchers who thought this third failure might be the nail in the coffin for LMTM were mistaken. Schneider explained that TauRx changed the statistical analysis plan for this latest Phase 3 trial before the data were locked. The revised analysis appears to be in response to subgroup analysis of the first Phase 3 AD trial, which claimed a positive effect in a small number of patients who were not on AChEIs. Presented at AAIC in Toronto, that earlier subgroup analysis was widely panned by statisticians for comparing apples and oranges: The drug sponsors compared people on LMTM monotherapy against the whole placebo group, which included those on AChEIs as well. At AAIC, experts explained that people who take AChEIs do so for a reason—their disease is worse and they are declining faster (see Schneider et al., 2011). Clinicians predicted that patients who are not taking AChEIs would decline more slowly owing just to their slow disease progression, not because they were taking LMTM.
This second AD trial provided an opportunity to test whether LMTM monotherapy was better than no therapy. Alas, Schneider showed comparisons between the 100 mg monotherapy group and the whole placebo group, i.e., again including people on AChEIs. He also compared the 4 mg LMTM/placebo “monotherapy” with those taking 4 mg LMTM in addition to AChEIs. “These are completely unfair comparisons,” said Suzanne Hendrix, president and CEO of Pentara Corp., Salt Lake City. “They have to compare within the same subgroups, namely LMTM monotherapy against placebos who are also not taking other therapy.”
This questionable comparison did show an effect as per Schneider’s presentation, but without a dose response, which in the world of clinical trials often means that something is not right. Both the 100 mg and 4 mg monotherapy groups declined more slowly than those taking placebo and AChEIs, but at exactly the same rate, making some experts wonder if they were simply the inherently slow responders. Over the 18-month trial, both monotherapy groups dropped about 2.5 points on the ADAS-Cog, 5.0 points on the ADL, and their left brain ventricular volume expanded by the same amount.
Lawrence Honig, Columbia University, New York, told Alzforum that the subgroup analysis was spurious, and that this trial, like the prior trial, showed no evidence to support any efficacy of the LMTM, nor any reason to proceed with further development of this drug. Aisen called the data meaningless. Others at the meeting said they were horrified at how the data had been sliced but requested their names not be used.
Unperturbed, Claude Wischik, CEO of TauRx, told Alzforum that he plans to abandon the higher doses and develop the 4 mg “placebo” dose. “We are making no further claim other than the data are sufficiently interesting to warrant a further study,” he said. Since 4 mg of LMTM also turns urine green/blue, it is not clear what would be used as a color placebo. Wischik said details of any new trial would be decided after consulting with regulatory agencies. Typically, sponsoring companies consult senior scientists at the FDA and EMA for input during the development process. Wischik said he had not yet discussed further development of LMTM with those agencies.
Hendrix did not believe the data warrants a new trial. “If there is evidence of a treatment effect for LMTM to support moving into another Phase 3 study, either overall or in a subgroup, it has not been presented publicly," she told Alzforum.—Tom Fagan
Much ‘Adu’ About a Little: Phase 1 Data Feeds the Buzz at CTAD
Let down by solanezumab’s demise, researchers at the Clinical Trials for Alzheimer’s Disease conference, held December 8-10 in San Diego, were in sore need of some good news. They got a taste of it, albeit small, with the release of new data from PRIME, the Phase 1b trial of the anti-Aβ antibody aducanumab. Biogen researchers presented results of a titration arm of the trial, in which gradually upping doses in ApoE4 carriers not only limited the amyloid-related imaging abnormality (ARIA) that signifies edema in the brain, but also cleared more brain amyloid than seen in participants on either of the two lowest doses of the drug. Participants in a long-term extension study also saw their brain amyloid load continue to shrink. In a separate bit of hopeful news, researchers from Genentech showed data from a Phase 1b trial of their anti-Aβ antibody, crenezumab. They reported that higher doses were safe and reaffirmed the company’s decision to boost the dose in a Phase 3 trial. Results from Phase 3 trials of both immunotherapies are expected in 2020.
Suzanne Hendrix of Pentara Corporation in Salt Lake City commented that in terms of their trial results and potential as therapies, solanezumab and aducanumab are polar opposites: Results from the aducanumab trial were tantalizing yet fraught with large error bars and small sample size, while the solanezumab trial yielded statistically solid results whose effect size was underwhelming.
The monoclonal antibody aducanumab latches onto oligomeric and fibrillar forms of Aβ, whereas solanezumab preferentially binds soluble monomeric Aβ. Phase 1 results of aducanumab in people with prodomal and mild AD were gradually offered up in tasty morsels at conferences over the past two years, dazzling researchers and investors. Findings suggested dramatic reductions in Aβ burden—in some cases to near-normal in people who began the trial with a brain full of plaques (see Mar 2015 conference news and Aug 2015 conference news). To the chagrin of some researchers, Biogen teased the field with exploratory findings indicating a cognitive benefit, and a post hoc data analysis indicating that this happened only in people whose brain amyloid had shrunk (see Sep 2016 news). The sword of Damocles hanging over this antibody was widely seen to be its dose-related increase of ARIA-E, particularly in ApoE4 carriers.
In hopes of managing this problem, researchers hypothesized that gingerly nudging up the dose might reduce the occurrence of ARIA-E in those patients. To investigate, they added a titration group to PRIME, along with eight ApoE4 carriers in a new placebo group. Over 12 months, 23 ApoE carriers received two doses of 1 mg/kg, four doses of 3 mg/kg, five doses of 6 mg/kg, and three doses of 10 mg/kg aducanumab, while eight ApoE4 carriers received placebo. At CTAD, Biogen’s Vissia Viglietta superimposed data from this titration arm on the previously published data from the 1, 3, 6, and 10 mg/kg groups, as well as the pooled placebo group of 48 participants. Amyloid-PET scans revealed that, similarly to the placebo and 1 mg/kg groups, no significant reductions in amyloid burden had occurred in the titration group at six months, by which time they had just started taking the 6 mg/kg dose. However, by 12 months, Aβ took a nosedive in the titration group, ending up between the continuous 3 mg/kg and 6 mg/kg groups. On the clinical dementia rating scale sum of boxes (CDR-SB)—an exploratory measure on which the trial was too small to draw conclusions—slowed decline in the titration group seemed on par with that seen in the 10 mg/kg group.
Importantly, ApoE4 carriers in the titration arm experienced less ARIA-E than did ApoE4 carriers who continuously received 6 or 10 mg/kg. Eight out of 23 patients in the titration arm (35 percent) had ARIA-E, compared with nine out of 21 (43 percent) and 11 out of 20 (55 percent) in the 6 and 10 mg/kg groups, respectively. The eight ARIA-Es in the titration group surfaced early in the trial, during the 3 or 6 mg/kg dosing segments. The researchers stressed that all these imaging abnormalities, along with their possibly associated symptoms such as headache, resolved within four to 12 weeks. Six of eight participants with ARIA-E continued treatment.
Based on this data, Biogen decided to employ this titration strategy in the Phase 3 studies of aducanumab. ApoE4 carriers will start at 1 mg/kg and titrate up to 6 mg/kg, while noncarriers will titrate up to 10 mg/kg, Biogen’s Samantha Budd Haeberlein told Alzforum.
In a separate presentation, Budd shared results from the first year of a three-year extension study, in which PRIME participants could choose to continue open-label treatment. People who had received 3 mg/kg, 6 mg/kg, or 10 mg/kg doses during the first 12 months stayed on the same dose for the extension, while those who had received 1 mg/kg switched to 3 mg/kg. People in the placebo group during the first year of the trial upped it to 3 mg/kg or to a titration, which started with two doses of 3 mg/kg followed by 6 mg/kg for the rest of the extension.
Scientists packing the hall let out a collective gasp when Budd flashed the 24-month amyloid-PET results, revealing a further drop in brain amyloid in each continuous dose group between 12 and 24 months. The trajectory of the amyloid curves for participants who had been in the placebo or 1 mg/kg groups had been nearly flat for those 12 months, but kinked downward after participants switched into 3 mg/kg for the extension.
Their clinical endpoints were less clear-cut. Performance on the CDR-SB leveled out between 12 and 24 months for people taking the 10 mg/kg dose, while it continued to decline in the 3 and 6 mg/kg groups. Decline in the 3 mg/kg group was slower than in the 6 mg/kg group. People whose dose went up in long-term extension, i.e., those who switched to 3 mg/kg from placebo or 1 mg/kg, declined slightly faster than did the continuous 6 mg/kg group.
Of 117 participants who started the extension trial, 26 discontinued treatment. Of those, 12 discontinued due to adverse events—the most common ones being falls, headache, and ARIA-E. Among patients who continued on their same dose, no new cases of ARIA-E cropped up in the extension portion of the trial. However, perhaps unsurprisingly, five of 29 “placebo switchers” and three of 17 people who switched from 1 mg/kg to 3 mg/kg developed ARIA-E. Seven of these eight incidents occurred in ApoE4 carriers, and four of the 12 adverse event-related drop-outs from the extension trial were due to these events.
At CTAD, many researchers expressed cautious optimism about this latest release of aducanumab results. Some wondered about potential effects of the trial’s staggered enrollment. The 1 mg/kg, 3 mg/kg, and placebo groups started first, followed by a 10 mg/kg group along with another placebo group. After ARIA-E started cropping up in them, the researchers added the 6 mg/kg dose and later the titration arm, each along with another placebo group. For each separate enrollment, participants were randomized 3:1 to active versus placebo groups. The 6 mg/kg group did not fall neatly in line with the dose-response curve for cognitive endpoints—landing in between the 3 mg/kg and 1 mg/kg groups on the MMSE at 12 and 24 months, as well as on the CDR-SB at 24 months. Noting that raters of cognitive tests, who were privy to the timeline, might make assumptions about which dose group a person belonged to, Hendrix asked whether this could skew assessors to score participants with later start dates higher if they thought they were on higher doses. Comparing cognitive scores among the placebo groups could partly allay such concerns, Hendrix told Alzforum (see Nov 2015 news).
Budd said that while investigators were indeed aware of the enrollment schedule, they were blinded to randomization to active treatment or placebo. She said investigators noted no obvious differences between the placebo groups but that because those groups were small, about eight patients each, the study was not powered to detect such differences. “Pooling the placebo groups was part of the prespecified analysis,” she told Alzforum. Several site investigators, including Larry Honig of Columbia University, New York, noted that with such small numbers of participants in each arm, they would like to see how individual patients performed over time, for example by way of spaghetti plots.
Jacking Up Crenezumab
With its own Phase 3 trial in the starting blocks and new results from Phase 1b, Genentech’s crenezumab program sits at a similar stage of development as aducanumab, though its Phase 3 program at present is smaller. Crenezumab—a humanized IgG4 monoclonal antibody—binds to oligomeric forms of Aβ. Because IgG4 antibodies exert little effector function, meaning they trigger fewer potentially destructive cytokine responses, researchers hope to be able to use higher doses without triggering neuroinflammation that may underlie ARIA-E.
At CTAD, Helen Lin, of Genentech in South San Francisco, presented results from a new Phase 1b dose-escalation study that the company is using to determine the best doses for its nascent Phase 3 trial, CREAD. Genentech tested the safety and tolerability of 40, 60, and 120 mg/kg doses after patients had received 15 mg/kg in a previous Phase 2 trial. The decision to jack up the dose stemmed from findings of a new drug-disease progression model, which accurately predicted cognitive decline the scientists observed in two Phase 2 studies. The simulation predicted that 60 mg/kg would produce the maximal treatment benefit without safety concerns.
Similar to the aducanumab Phase 1b design, the crenezumab dose-finding study staggered enrollment. The first cohort was randomized 5:1 to 40 mg/kg crenezumab or placebo, infused four times over 13 weeks, with safety and pharmacokinetic measurements at weeks five and 13. The other two dose groups (and corresponding placebo groups) started five and 10 weeks later. Following each cohort’s 13-week placebo-controlled phase, participants transferred into an open-label, long-term extension program. People on placebo switched to the dose taken by the treatment group in their cohort (except people in the 120 mg/kg cohort switched down to 60 mg/kg for the extension).
Lin described safety data for the 40 and 60 mg/kg doses for the placebo-controlled portion of the trial. She reported that none of the study’s 52 participants—mild to moderate AD patients ranging from 50 to 90 years of age—experienced ARIA-E. Six participants did have ARIA-H (hemosiderin), generally considered a less worrisome, asymptomatic abnormality. There were no severe adverse events. The adverse events that did occur tended to be mild and did not force an end to treatment. In the 60 mg/kg group, the serum concentrations of the antibody quadrupled over those observed in people who received 15 mg/kg infusions in the Phase 2 trial, as expected.
Lin told the crowd that Genentech is moving forward with its 60 mg/kg dose in a Phase 3 trial of 750 people with prodromal to mild AD, which is slated for completion in 2020.
Active Immunization: A More Cost-Effective Shot at Goal?
If aducanumab, solanezumab, or crenezumab succeed in Phase 3, most researchers agree that the earlier treatment begins, the better the chances of slowing neurodegeneration. Alas, an early start to treatment would mean years of costly infusions of a biologic therapy. Cynthia Lemere of Brigham and Women’s Hospital in Boston was impressed by the results from the PRIME aducanumab trial, but questioned whether long-term treatment would be cost-effective or even accessible to the common person. Lemere speculated that should an Alzheimer’s vaccine exist, plenty of middle-aged people, especially those with a family history of the disease, would rush to get those shots.
The standard-bearer in this area is Novartis’s CAD106, which is in a Phase 2/3 trial expected to last until 2023. But several other Aβ vaccines are coming up in its wake, and one was reported at CTAD.
Pedro Pesini of Araclon Biotech in Zaragoza, Spain, presented safety and immunogenicity data from a Phase 1 study of ABvac40, a vaccine directed against the C-terminus of Aβ40. The study enrolled 24 patients with mild to moderate AD who received three monthly subcutaneous injections, either of the vaccine (16 patients) or placebo (eight patients). Pesini reported no significant differences in adverse events between the treatment and placebo groups. Most people in the treatment group generated antibodies against the vaccine, with plasma titers ramping up more after each injection. These antibodies were still detectable in the plasma a year later, Pesini said. —Jessica Shugart
Tau PET in Down’s: Unique Patterns Among Alzheimer’s Types and Stages
By the time memory frays, the brains of people with Alzheimer’s disease are already infested with Aβ plaques, but the growth of tau tangles tracks with worsening symptoms. With tau PET, researchers now have a tool to watch this happen, and at the Clinical Trials on Alzheimer’s Disease (CTAD) conference, held December 8-10 in San Diego, they discussed their latest data to see how close the field is to using tau PET in human therapy studies. Showing where tau tangles are in people with early versus late-onset AD, Down’s syndrome (DS), and among participants in secondary prevention trials, they compared the differences in tangle distribution. The extent to which tau pathology pushed into the neocortex differed among forms of the disease, but the requirement that Aβ pathology be there to unleash tau’s toxic spread was consistent between studies. Researchers grappled with how best to wield this powerful imaging tool in clinical trials.
A new kid on the block compared to amyloid-PET imaging, tau PET has allowed researchers to confirm during life Braak’s postmortem neuropathology finding that tangle pathology spreads out of the medial temporal lobe into the neocortex as AD progresses (see Braak and Braak, 1991). When combined with the more established amyloid-PET or measurements of CSF Aβ, tau imaging has also implicated elevated Aβ deposition as a prerequisite for tau’s pathological spread into the neocortex (see Mar 2016 news; May 2016 news). Trialists hope to harness this biomarker to select participants and perhaps measure outcomes, but CTAD showed they still have a long way to go before they can lean on tau imaging with confidence. “It’s still early days,” said Michael Schöll of Lund and Gothenberg universities in Sweden.
At CTAD, Reisa Sperling of Brigham and Women’s Hospital in Boston presented the first tau PET data from A4, the ongoing secondary prevention trial testing solanezumab in cognitively normal people with elevated Aβ deposition (see Jan 2013 news). Thus far, nearly 250 tau PET scans have been performed on a subset of the more than 800 participants already randomized across 67 sites in the United States, Canada, Australia, and Japan, Sperling said. While Sperling stressed that no consensus yet exists for a “tau-positive” threshold, she reported that 56 percent of the cohort had standardized uptake volume ratios (SUVRs) of the tau tracer AV-1451 above 1.2, which she classified as elevated neocortical tau.
Sperling reported that even within this presymptomatic cohort, the toxic relationship between Aβ and tau appeared to be firmly established. People at the high end of the Aβ spectrum had more tau pathology in the entorhinal, inferior temporal, and inferior parietal cortices, and precuneus. Sperling told Alzforum that, to her surprise, tau accumulation among A4 participants ranged widely, from primarily medial temporal tangles in some participants to extensive neocortical pathology on par with later Braak stages in others. Sperling referred to people at the high end of the tau spectrum as possible “bell-ringers,” people whose PET data is loudly signaling that they are on the edge of cognitive decline. She said that tau imaging in this cohort may help researchers home in on an ideal treatment window for trials—a time just prior to clinical expression of the underlying disease process—and also to investigate how Aβ-targeted therapies alter the course of tau pathology.
Michael Rafii of the Alzheimer’s Therapeutic Research Institute at the University of Southern California and the University of California, San Diego, presented data from the first tau PET imaging in people with DS. Owing to their extra copy of chromosome 21, which hosts the APP gene, people with DS develop AD pathology at a young age. Researchers recently added tau PET imaging to the Down’s Syndrome Biomarker Initiative (DSBI), a three-year longitudinal study in people with DS (see Dec 2012 news; Rafii et al, 2015). In addition to cognitive tests, as well as amyloid-PET, structural MRI, and FDG-PET imaging acquired previously in the study, the researchers ran tau PET scans on nine participants, averaging 48 years of age, at the two-year mark. These nine displayed no cognitive symptoms of AD yet, though six of them had elevated Aβ at baseline.
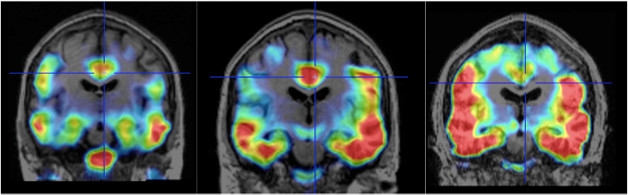
At CTAD, Rafii reported that, similar to presymptomatic people in A4, tau accumulation in people with DS was associated with elevated Aβ deposition. The three participants without elevated Aβ did not have tau deposition beyond the medial temporal lobe, whereas those with elevated Aβ displayed the typical “batwing” distribution of neocortical tau (see image below). What’s more, tau accumulation correlated with waning cognitive performance between baseline and two years, as well as with atrophy in the medial temporal lobe.
Batwing in Down’s. As tau spreads from the medial temporal lobe into the neocortex, a batwing pattern of tau tracer uptake appears. Scans were conducted on a 48-year-old (left), 50-year-old (middle) and 52-year-old (right) person with DS prior to onset of AD symptoms. [Images courtesy of Michael Rafii, ATRI and UCSD.]
“This fits with the idea that AD biomarkers behave similarly in people with DS as they do in other people with AD,” Rafii told Alzforum. “As such, people with DS are being included in AD prevention trials such as the ACI-24 in DS study," he said.
Lund University’s Schöll compared tau accumulation patterns between early and late-onset AD in the Swedish Biofinder Study. More than 1,600 people participate in this longitudinal study, which aims to identify early biomarkers for a range of neurodegenerative diseases, including Alzheimer’s. Schöll and colleagues conducted tau scans in 17 healthy controls, 16 people with EOAD (defined as onset before age 65), and 17 with LOAD (onset after age 65). Neither AD group harbored familial AD mutations. Unlike Sperling and Rafii’s cohorts, participants in this tau PET sub-study were symptomatic (with average MMSE scores of 21.4 in the EOAD group and 20.6 in the LOAD group) and thus expected to have more advanced tau pathology. Schöll reported that tau distribution was markedly different between the two groups. People with EOAD had tangles in the medial temporal lobe and well outside of it—extending into the posterior, parietal, and occipital cortices. On the other hand, the bulk of tau deposition in people with LOAD remained closer to its origins in the medial temporal lobe, spreading from there into inferior and lateral temporal lobes, but largely avoiding posterior areas.
Schöll speculated that the Aβ-dependent process that drives tau into the neocortex could somehow work differently in EOAD and LOAD even though findings regarding differences in Aβ accumulation between the two groups have been inconsistent. Tau deposition more closely overlapped with atrophy in people with EOAD than LOAD. “The diseases differ both in their relationship between Aβ and tau, and also in the effect of tau on atrophy,” Schöll told Alzforum. Whether these differences relate to the more aggressive nature of early onset disease remains to be seen, he said. Schöll added that clinicians should consider these differences between early and late-onset AD when using tau PET as a selection or monitoring tool in trials, once this pattern has been established further.
How does tau PET compare to measurements of cerebrospinal fluid tau and phospho-tau as a diagnostic or disease progression biomarker? Discussing a poster by Niklas Mattsson, also of Lund, Schöll reported that while both worked as diagnostic markers, tau PET far outperformed CSF total tau or phospho-tau in tracking progression. The researchers conducted tau PET and measured CSF tau/p-tau in 29 people with AD, eight people with MCI, and 17 healthy controls. They found strong correlations between the two modalities in people with low amounts of tau pathology in classic Braak regions, while the association crumbled in people in the later stages of disease, who had a heavy tau burden. One possible interpretation of this finding is that CSF levels of tau and p-tau bump up in the early stages of the disease but then plateau as tau accumulates further, Schöll said, adding that beyond early stages of the disease, tau PET appears to be a far more accurate measure of disease progression.
Filling in for Lund’s Oskar Hansson, who heads the Swedish Biofinder Study but was unable to attend CTAD, Schöll also presented tau imaging data from other neurodegenerative diseases. Originally developed to detect the combined four-repeat and three-repeat tau tangles that occur in AD, the AV-1451 tracer has come up short in other tauopathies that consist of pure 4R or 3R tau. Schöll described recently published data indicating that people with the 4R tauopathy progressive supranuclear palsy (PSP) took up the tracer only in their basal ganglia, an area where background uptake in healthy controls is high. Although tracer uptake in the basal ganglia correlated with PSP severity, Schöll said the high background uptake limited the usefulness of the tracer in people with this disease. There was no uptake in regions of the cortex known to harbor tau aggregates in people with PSP (see Smith et al., 2016).
Results were slightly more encouraging in people with corticobasal degeneration, another 4R tauopathy. Neurodegeneration of the motor cortex and subcortical motor tracts tends to predominate on one side of the brain in CBD, and Schöll reported that tau tracer uptake was indeed highest in those regions on the affected side in six people with the disease. Tracer uptake on the less affected side was much lower, partly overlapping with uptake in healthy controls. Why AV-1451 was taken up in CBD but not PSP is unclear, Hansson told Alzforum by phone later, although he speculated that the tracer might detect pure 4R tau if accumulation surpassed a certain threshold. He stressed that these initial studies are exceedingly small, and much larger series are needed to determine if and how tau imaging is useful in neurodegenerative diseases other than AD. —Jessica Shugart
Emerging Alzheimer’s Therapies Test the Waters at CTAD
Beyond keynote speeches and eagerly awaited immunotherapy data, many presentations at the Clinical Trials on Alzheimer’s Disease (CTAD) conference, held December 8-10 in San Diego, detailed findings from therapies in the earliest stages of development. These were tested in small groups of participants, some without placebo controls. Here are a few highlights from trial tidbits reported at the conference.
Philip Scheltens of VU University Medical Center in Amsterdam presented data from a Phase 2a trial of VX-745, aka Neflamapimod. This p38MAPK inhibitor is proposed to reduce harmful inflammation, boost microglial phagocytosis, and improve neuronal plasticity. Sixteen people in Europe with MCI or mild AD received either 40 mg or 125 mg of VX-745 twice daily for 12 weeks. Fifteen (eight in the 40 mg group and seven in the 125 mg group) completed the trial and were evaluated at the VU Medical Center. The drug was well-tolerated and caused no severe adverse events, Scheltens said.
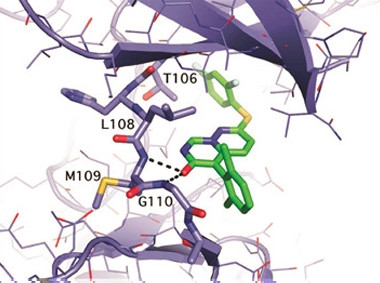
Kinase Blocker.
Crystal structure of VX-745 (green) bound to p38 MAPKα (purple). [Courtesy of John Alam.]
Researchers evaluated the primary outcome measure—reduction in Aβ deposition—via quantitative PET imaging. This method, which continuously measures PiB in the brain throughout a 90-minute scan, is more sensitive and less prone to variability than standard amyloid-PET scans, which take a briefer measurement at a specific time, usually 60 to 90 minutes after the tracer has been injected into the blood stream. Because of the tighter variability of this PET method, the researchers decided ahead of time that an amyloid reduction of more than 7 percent would constitute a bona fide response to the drug, while a 3 percent to 7 percent reduction would be considered a partial response. They found that in the 40 mg group, three people had a full response, two had a partial response, and three did not respond. In the 125 mg group, only one out of the seven had a full response. Interestingly, that person had a plasma level of the drug on par with people in the 40 mg group. All four full responders had plasma VX-745 exposure below 90 nghr/ml.
Scheltens reported that the four full responders also had had the lowest baseline levels of amyloid deposition. He concluded that the lower, 40 mg dose was better for reduction of amyloid, and that three months of treatment was perhaps insufficient to reduce amyloid deposition in those who started out with high baseline levels. Scheltens could not explain these paradoxical dose findings, although he offered that it is possible that the higher dose may go too far in shutting down inflammatory responses needed to mop up Aβ.
In a secondary outcome measure, the Wechsler Memory Scale test, participants in both dose groups improved throughout the trial. However, Scheltens acknowledged that because this small trial had no placebo group, it would be difficult to rule out that practice effects were at play.
Separately, John Alam of EIP Pharma in Cambridge, Massachusetts, reviewed results from a U.S.-based CSF analysis study with the same basic design, though it was shorter—six weeks rather than 12. Sixteen people with MCI or mild AD were slated to receive either 40 mg or 125 mg of VX-745 twice daily. However, after the trial began, the FDA introduced a limit on dosing, to keep plasma VX-745 levels tenfold below the level causing toxicity in long-term animal studies. Because the drug sponsors expected plasma VX-745 might exceed that limit in people taking the high dose, they suspended that arm after only three people had been enrolled and one had completed the study. Alam told Alzforum that because the European Union interprets the toxicity differently, the high-dose arm could be completed in Amsterdam.
The U.S. study measured memory using the Hopkins Verbal Learning Test–Revised (HVLT-R), a test that affords little improvement with practice. This was evident by minimal improvement of most participants in two tests before they took their first dose, Alam said. In the HVLT-R, participants are asked to remember as many words as possible from a list of 12, either immediately or 20 to 25 minutes after hearing them. Seven and five of the eight participants on 40 mg VX-745 improved significantly in the immediate and delayed recall scores, respectively, at the end of the trial.
Alam reported analysis of CSF taken from the eight participants who received the 40 mg dose of VX-745 and the one who completed the 125 mg dose regimen before that was stopped. Since that person’s plasma VX-745 was on par with those in the 40 mg dose, the researchers pooled the data. Because VX-745 is thought to tamp down neuroinflammation, the researchers attempted to measure the CSF concentrations of nine cytokines, but ultimately only one—IL-8—was consistently detectable. CSF levels of this pro-inflammatory cytokine significantly dropped in three participants who had the highest plasma levels of VX-745, all above 90 nghr/ml, suggesting that the drug does have some anti-inflammatory effects, Alam reported. These same three participants with less CSF IL-8 also had significantly elevated CSF levels of Aβ38 and Aβ40 relative to their baselines, with a similar trend for Aβ42
While the PET and CSF analysis were undertaken in different patients, at different centers, Alam told Alzforum that it does not appear VX-745 reduces amyloid plaque via an anti-inflammatory effect (i.e., by reducing IL-8 levels). The PET responders in Amsterdam all had plasma drug exposures below 90 nghr/ml, while those in the United States whose plasma IL-8 fell all had drug exposure above that level. Alam said the data is in keeping with preclinical studies suggesting VX-745 has interleukin signaling effects mediated by inhibition of p38α, and interleukin production effects mediated by inhibition of p38β. The drug’s IC50 for the former is less than for the latter, Alam said. This may explain why lower doses of the drug might preferentially reduce Aβ—higher doses could interfere with its clearance. Suppressing p38α may attenuate BACE cleavage of APP, and slow production of Aβ42 (see Schnöder et al., 2016). Alam thinks this may be how the drug improves cognition, because that might reduce Aβ toxicity at the synapse. The researchers hope to further test the drug’s effects on Aβ burden and cognitive improvement in a larger, placebo-controlled trial, Scheltens said.
Ruolun Qiu of Pfizer in Cambridge, Massachusetts laid out data from PF-06648671, a small molecule designed to clamp down on γ-secretase’s production of amyloidogenic peptides from APP, while sparing processing of other crucial substrates, such as Notch 1. Qiu presented safety and CSF Aβ data from single- and multiple-dose studies.
In a single-dose study, 22 healthy participants, ages 18 to 55, received one oral dose of 150 mg or 300 mg of PF-0664871, or a placebo, and their CSF was monitored continually for 36 hours via a lumbar cannula. Aβ40 and 42 levels reached a nadir at 16 hours—dropping by an average of 23 percent and 39 percent, respectively, in response to the 300 mg dose. In a multiple-ascending-dose study on healthy volunteers in the same age group, six cohorts of 10 (eight active, two placebo) participants received daily doses ranging from 4 mg to 360 mg of the modulator, or placebo, for 14 days. On day 14, the researchers observed a dose-dependent reduction in Aβ42—by 14, 43, 59, and 65 percent for the 40, 100, 200, and 360 mg doses, respectively. Aβ40 also dropped dose-dependently, but by lesser amounts, resulting in a reduction in the Aβ42/40 ratio just as with the single dose.
On the other hand, shorter peptides shot up in response to treatment. Aβ37 rose by 335 percent and 372 percent after 14 days on 200 mg and 360 mg doses, respectively, while Aβ38 rose by 46 percent after 14 days on the 200 mg dose. Some of these peptides are considered non-amyloidogenic, but the full range of Aβ species has not been exhaustively studied. Despite a dramatic shift in the length of certain Aβ peptides, treatment did not affect the concentration of total Aβ, Qiu said. These findings suggested to the researchers that the drug could limit production of toxic Aβ species while otherwise preserving γ-secretase function. Qiu reported that the drug was safe and well-tolerated at the tested dose range, and that the most common adverse events—headache, dizziness, and nausea—were likely due to the lumbar puncture procedure. The safety profile also looked good in a third study in healthy elderly people, ages 65-85, who received 200 mg doses for two weeks, Qiu said.
Another drug aimed at altering neuronal activity, AGB101, is poised to enter Phase 3. An extended release version of the anti-epileptic drug levetiracetam, AGB101 is proposed to dampen the neuronal hyperactivity in the hippocampus that occurs in people with mild cognitive impairment. In a Phase 2 study, the drug appeared to do exactly that, and people performed better on memory tests (see Mar 2015 news). At CTAD, Richard Mohs of AgeneBio in Baltimore and colleagues reported that following a positive meeting with the FDA, the company will move forward with Phase 3 trials to test the drug’s effects on cognition. Starting in mid-2017, they will randomize 830 people with MCI due to AD to receive placebo or 220 mg of AGB101 for 78 weeks. The primary outcome measure will be improvement on the clinical dementia rating scale sum of boxes (CDR-SB). Participants will also undergo other cognitive tests and structural MRI to track the drug’s effects on neurodegeneration. AgeneBio has spent 2016 preparing and raising money for this Phase 3 study (see slide deck).
A Placebo-Free Space for Therapy Trials?
Tackling AD from a metabolic angle, John Didsbury of T3D Therapeutics in Research Triangle Park, North Carolina, presented findings from a Phase 2a study of T3D-959 in patients with mild to moderate AD. The hope is that this small-molecule agonist of PPAR delta/gamma nuclear receptors will improve waning metabolism in the AD brain by increasing insulin sensitivity. Thirty-six participants were randomized to receive 3, 10, 30, or 90 mg daily oral doses of the drug for 14 days. There was no placebo group.
On treatment day 14, FDG-PET imaging indicated dose-dependent changes compared to baseline in cerebral glucose metabolism in brain regions typically affected by AD. For the most part, the study measured regional to whole-brain ratios of glucose metabolism, which went down in a dose-dependent manner. To Didsbury, this indicated that the drug penetrated the brain and engaged its target.
On a secondary outcome measure, the ADAS-Cog11, 17 of the 32 participants available for testing at day 14 reportedly improved by at about one point over baseline, while 12 and nine participants improved by two or three points, respectively. Improvements held steady when participants were re-tested one week later. Despite the small number of participants on whom to base his conclusions, Didsbury claimed the dose response differed by ApoE4 genotype. Among non-carriers, those in the lower-dose groups improved over the course of the study, while those in the highest-dose group got worse. Carriers, on the other hand, had a more classic dose response, improving more at higher doses, said Didsbury. He said that researchers will take ApoE4 status into account in future, placebo-controlled studies. Others have noted that two- to three-week cognitive data, much less genetic stratification, in small, short trials, without a placebo group, carry little meaning.
A combination of repurposed drugs also was reported at CTAD, when researchers from Pharnext in Paris presented data from a single-blind, exploratory Phase 2 trial of PXT-864. In keeping with the company’s modus operandi of putting previously approved drugs to new use, this regimen consists of the GABA-B receptor agonist baclophen (a muscle relaxant) and the small molecule acamprosate (approved to treat alcohol dependence). Pharnext researchers propose that the cocktail could help balance inhibitory and excitatory synaptic signals skewed by Aβ oligomers. The trial started as a 12-week pilot study called PLEODIAL I. Forty-five people with mild AD received one of three combination doses twice daily (0.4 mg acamprosate and 6 mg baclofen, 1 mg acamprosate and 15 mg baclofen, or 20 mg acamprosate and 12 mg baclofen). There was no placebo group. Participants took the combination for two four-week stints, with a four-week placebo break in the middle. Then, in a 24-week extension study called PLEODIALII, 39 volunteers continued the treatment and had the option of taking 5 mg donepezil along with PXT-864 during the last 12 weeks of the extension.
At CTAD, René Goedkoop of Pharnext reported that over the course of the 36-week trial, participants in each dose group declined more slowly—by 0.6 to 0.9 points—on the ADAS-Cog11 than did published, historical placebo groups. However, in other contemporary treatment trials, too, placebo groups sometimes decline more slowly than historical controls. When the researchers removed participants who took donepezil during the last 12 weeks of the trial from the analysis, they found that people in the lowest-dose group actually declined faster than historical placebos—by an average of 2.3 points— over the course of the trial. Removing donepezil-takers did not affect the results in mid- or high-dose groups, but the findings were confounded by an uneven distribution of donepezil takers: Seven of 13 participants on the low dose, compared to only two of 14 in the middle and one of 13 on the high dose, opted to take donepezil.
On a poster, Karim Bennys of the University Hospital in Montpellier, France, presented results of event-related potential (ERP) recordings of three participants in each dose group of the PXT-864 trial. The latency and amplitude of ERPs in response to a task are thought to reflect working memory or decision-making. Both measures shot up during the treatment phases relative to baseline and the interim placebo phase, in people on the low and middle doses. To Bennys, this indicated that the drug may have a neurophysiological effect. At 36 weeks, the three people on the low dose had the largest improvements in ERP latency and amplitude; however, all three of them also took donepezil during the last 12 weeks of the trial (those on mid and high doses did not take donepezil). After the first four weeks of the trial, the six participants on the highest combination dose had improved in ERP latency and amplitudes, however, they declined following the next treatment phase.
Goedkoop told Alzforum that it is difficult to say whether this group performed more poorly due to the absolute dose, or to the different ratio of acamprosate to baclofen in that group, which was 5:3 rather than 1:15. The researchers aim to tease out these dose effects, and the interaction with donepezil, in future double-blind, placebo-controlled Phase 2 trials, Goedkoop said.—Jessica Shugart

Comments
No Available Comments
Make a Comment
To make a comment you must login or register.