When more than 30,000 registrants gathered at the San Diego Convention Center for the Society for Neuroscience annual meeting November 12-16, they exchanged their latest data on the basic science on Alzheimer’s disease. Topics ran the gamut from protein propagation and inflammation to the hunt for plasma biomarkers and potential therapeutics. Alzforum reporter Madolyn Rogers brings you highlights.
Astrocytes and Exosomes Implicated in Protein Propagation
Many researchers now believe that misfolded proteins spread from cell to cell across the brain, corrupting normal proteins as they go, yet exactly how this propagation would happen remains unclear. At the Society for Neuroscience annual meeting, held November 12-16 in San Diego, scientists identified specific mechanisms that may be involved in the spread of pathological proteins in Parkinson’s and Alzheimer’s diseases. They argued that astrocytes transmit α-synuclein aggregates by cell-to-cell contact; for tau, they argued that its trans-synaptic spread depends on the secretory vesicles known as exosomes. In addition, researchers debuted a new method for following tau aggregates over time in living mouse brain.
“Up until a few years ago, we did not understand how Parkinson’s disease progresses at the molecular level,” noted Alice Chen-Plotkin of the University of Pennsylvania, who moderated an SfN press conference on Parkinson’s research. “Now, we may be starting to discover how the disease worsens, which could give insights into ways to slow it down or stop it,” she wrote to Alzforum.
Numerous studies have found that misfolded proteins can seed aggregation of normal protein throughout animal brains, and that such seeds may even be transferred from one person to the next in tissue transplants (see Apr 2015 conference news; Jan 2016 news). Many proteins fold into specific shapes, or “strains,” which propagate in vitro and give rise to distinct pathologies and symptoms in animal models (see Sep 2013 news; Jun 2015 news; Nov 2016 news). However, some researchers have pointed out that existing data fall short of demonstrating a causal role for protein propagation in human disease, or showing how proteins travel through tissue (see Apr 2016 webinar).
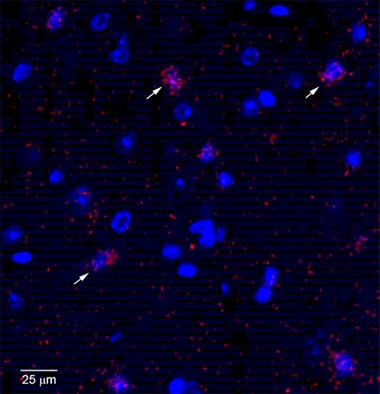
Glial Indigestion.
Cultured astrocyte (green) has ingested recombinant α-synuclein (red), which then accumulates around its nucleus (blue). [Courtesy of Jinar Rostami and Anna Erlandsson.]
Researchers led by Anna Erlandsson of Uppsala University, Sweden, working with Laurent Roybon of Lund University, also in Sweden, wondered whether astrocytes might aid and abet the migration of pathological proteins. Some recent studies have found that these glial cells pass misfolded prion protein to neurons (see Hollister et al., 2015; Victoria et al., 2016). At an SfN press conference, Jinar Rostami in Erlandsson’s group made the case that astrocytes transmit α-synuclein aggregates as well.
The researchers tested for protein spread using astrocytes derived from human pluripotent stem cells. Some researchers have pointed out that cultured astrocytes resemble immature or reactive cells more than mature brain astrocytes. However, the iPSC-derived astrocytes expressed markers for mature astrocytes, such as S100β, but low levels of proteins that mark reactive astrocytes, such as GFAP, Rostami told Alzforum. Rostami added recombinant monomeric or oligomeric α-synuclein to these cultures. Astrocytes gobbled up the proteins. They quickly degraded the monomers, but oligomeric α-synuclein was a different story. It lingered inside cells, passing through lysosomes to end up in the trans-Golgi network around the nucleus (see image above). After six days, astrocytes containing α-synuclein aggregates betrayed signs of cellular stress, including fragmented mitochondria and swollen endoplasmic reticulum. The cells began to pass the α-synuclein aggregates off to other astrocytes, as though playing a cell-culture game of “hot potato.” Time-lapse microscopy revealed instances of astrocytes making direct contact with neighboring cells to transfer labeled aggregates. In other cases, cells formed long, thin tunneling nanotubes to reach more distant cells (see Gerdes et al., 2013). Aggregates traveled down these tubes. Adding an inhibitor of cytoskeletal reorganization to the cultures to suppress contacts between cells cut α-synuclein transfer in half, Rostami reported in San Diego.
Does this happen in the Parkinson’s disease brain? This is unknown, but Rostami plans to look in vivo in mouse models. Meanwhile, she believes α-synuclein released by dying neurons in the midbrain of Parkinson’s patients could be taken up by astrocytes there and spread further. Erlandsson noted that the data may have therapeutic implications. “If we could stimulate degradation of α-synuclein aggregates by astrocytes, it could reduce spreading,” she told Alzforum. Other researchers consider this a challenging proposition since at least in neurons, α-synuclein resists autophagy (see Apr 2013 news).
That said, one potential treatment being tested in Parkinson’s trials, the cancer drug nilotinib, enhances autophagic removal of the protein by blocking Abl, a kinase that protects synuclein from degradation (see Nov 2015 conference news).
Speaking at the same press conference, Collin Challis, working with Viviana Gradinaru of the California Institute of Technology, Pasadena, reported that α-synuclein deposits added to the intestines of rats stimulate misfolding of endogenous α-synuclein there. Endogenous, but now misfolded, synuclein then migrates up the vagal nerve to the brainstem and midbrain, where it seems to affect behavior. Challis’ findings dovetail with the hypothesis that α-synuclein aggregation can start in the gut in Parkinson’s disease, and from there propagate to the brain (see Jul 2011 news series; Oct 2016 news; Holmqvist et al., 2014).
From Gut to Brain?
Tissue clearing reveals rare α-synuclein fibrils (green) in the endothelial cells (nuclei shown in blue) of the gut lining, and their three-dimensional relationship with neurons (red) of myenteric plexus ganglia and astrocytes (white). [Courtesy of Collin Challis and the Gradinaru Group at Caltech.]
Challis and colleagues injected preformed fibrils of recombinant α-synuclein into the stomach and gut lining of 22 adult wild-type mice, with another 19 receiving control saline/BSA injections. Within one week, the former developed gastrointestinal problems, defecating more than control mice. This correlated with the accumulation of endogenous α-synuclein in the gut lining, as seen by staining with an antibody that detects α-synuclein phosphorylated at serine 129, but not the recombinant preformed fibrils, which remain unphosphorylated (see image above). Phosphorylation at S129 promotes oligomerization and probably does not occur after fibrils form, Gradinaru told Alzforum (see Volpicelli-Daley et al., 2011; Samuel et al., 2016). Fibril formation in the gut peaked at three weeks, then returned to normal by two months, and gastrointestinal dysfunction followed the same time course. Meanwhile, endogenous α-synuclein aggregates appeared in the brainstem by three weeks, and the midbrain by two months. At this later time point, the mice began having trouble turning around on a vertical pole, indicating muscle weakness or loss of motor control. The results support the idea that the propagation of misfolded α-synuclein from gut to brain causes disease symptoms, Challis said. He did not address whether astrocytes in the brain or the enteric plexus played any role in the spread of α-synuclein.
Once protein aggregates have reached neurons, how might they travel among them? Some recent research suggests that exosomes smuggle proteins out of one and into another (see Dec 2014 conference news; Oct 2015 news). In San Diego, Yipeng Wang of the German Center for Neurodegenerative Diseases (DZNE), Bonn, presented evidence supporting the idea that tau migrates in this fashion. Working with Eckhard and Eva-Maria Mandelkow at DZNE, Wang first characterized exosomes secreted by primary cortical neurons cultured from wild-type rats. He found that these exosomes contained full-length tau that remained largely unphosphorylated. Tau likely rode inside the vesicles, rather than on their surface, because proteinase K failed to digest the protein when added to intact exosomes.
The researchers then examined whether these exosomes could transfer tau between cells. Wang collected exosomes secreted by a mouse neuroblastoma cell line that expressed tau labeled with GFP, and added the exosomes to two-chambered microfluidic culture devices containing mouse hippocampal neurons. Each such neuron extended an axon through a microgroove to contact the neuron in the adjoining chamber. When Wang added the exosomes after four days of microfluidic culture, before the neurons had formed synapses, no tau passed to their neighboring cells. After 11 days in culture, however, once synapses had formed, human tau appeared in the distant neuron, indicating that functional synapses are required to transfer tau. Naked tau from sonicated exosomes added directly to the chamber did not transmit to the neighboring cell, suggesting that intact exosomes are required. Furthermore, Wang reported that depolarization of the cultured neurons stimulated exosome release, suggesting these vesicles are shed in some sort of regulated physiological process. Wang did not speculate on why that might be.
Does this exosomal transport play a role in transmitting aggregates of tau? Wang and colleagues expressed an aggregating form of tau, DK280, in the mouse neuroblastoma cells, and found that the aggregates passed among them, again via exosomes. This experiment did not use hippocampal neurons in the microfluidic chamber.
Could neurons from people do the same? To begin to test this, the researchers isolated exosomes from the cerebrospinal fluid of Alzheimer’s patients and from healthy controls. The exosomes from patients contained tau oligomers. When added to the mouse neuroblastoma cells that expressed DK280 tau, the human exosomes promoted tau aggregation. Exosomes mediate the spreading of tau, Wang concluded.
Aggregated Aβ, as well, may travel in exosomes. Working with Martin Hallbeck of Linköping University, Sweden, Maitrayee Sinha isolated exosomes from postmortem Alzheimer’s brain tissue. Sinha reported in San Diego that these exosomes contained more oligomeric Aβ, as judged by binding to conformation-specific antibodies, than those from age-matched control tissue. When she added these exosomes to human neuronal cultures generated from induced pluripotent stem cells, the Aβ oligomers were taken up and passed to neighboring cells. Neurons containing the aggregates began to die. The researchers were able to prevent Aβ transfer either by inhibiting endocytosis or by silencing genes involved in exosome production and secretion, demonstrating the key role of exosomes in this process.
A Window on Live Aggregation
Because following protein aggregation and propagation in vivo is technically challenging, most of the data in this line of research come from in-vitro studies. That said, Sarah DeVos and Brad Hyman of Massachusetts General Hospital, Charlestown, have devised a way to follow tau aggregates in the living brain. At SfN, DeVos described how she modified a fluorescence resonance energy transfer (FRET) assay developed by Marc Diamond at the University of Texas Southwestern Medical Center, Dallas (see Sep 2013 news). She incorporated a mutant human tau repeat domain labeled with cyan fluorescent protein along with another one labeled with yellow fluorescent protein in the same viral vector. DeVos tested the vector in primary neurons, and found that as predicted, transfected neurons expressed equal amounts of both types of labeled tau repeat. When the repeats aggregated, they produced the FRET signal.
The researchers then injected the vector into layer II-III neurons in the cortices of adult Tg4510 mice, which express human mutant P301L tau. Imaging the neurons through a cranial window, they saw the formation of tau aggregates in cells that took up the vector (for a three-dimensional visual of this, see the video clip below). With this system, the researchers can now track the same neuron over months, DeVos told the audience. She wants to study the fate of transfected neurons, for example by analyzing their gene expression to see how the presence of tau aggregates alters them. An audience member pointed out that the FRET assay merely detects aggregates, not necessarily true fibrils. DeVos said she will use electron microscopy to confirm the nature of the aggregates. She also intends to track whether aggregates are transported down axons, although the spatial limitations of the cranial window will prevent directly seeing transfer to distant projection areas.—Madolyn Bowman Rogers
Knock-In Alzheimer’s Mice Catch on More Broadly in the Field
Scientists agree that mouse models of Alzheimer’s only partially recapitulate the disease and, more ominously, that their strong overexpression of human AD genes renders them prone to artifacts. In 2014, the group of Takaomi Saido of RIKEN Brain Science Institute, Wako, Japan, generated three strains of mice with mutant human APP knocked into the endogenous locus, resulting in normal expression levels of the protein. Since then, Saido has lobbied the field to study his mice and today, nearly 200 laboratories around the world do. So what do scientists know by now? Do these mice better model Alzheimer’s disease? What are their limitations?
A symposium at the Society for Neuroscience annual meeting, held November 12-16 in San Diego, tackled these questions. Scientists who work with the mice presented their latest data, other groups who use them were in the audience, and an enthusiastic crowd made for lively debate during a panel discussion. Overall, researchers remained divided for the time being on whether the knock-ins model AD more faithfully than do overexpression mice, especially on aspects where data differ between the two. Some investigators consider the very mild behavioral phenotype of the knock-ins to be a limitation, while others complained that their pathology develops slowly, hence experiments take longer. However, most scientists agreed that the knock-ins represent a valuable alternative to current models and that their increasing use will settle these open questions.
Amyloid Unleashed. Three month-old APP knock-in mice (left) have no amyloid deposits (green) in the cerebral cortex. Without the amyloid protease kallikrein 7 (right), they do, and gliosis to boot (red). [Courtesy of Taisuke Tomita.]
Pros and Cons of Knock-In Mice
What’s wrong with overexpression models? While they have generated many insights into the disease, researchers worry that insertion of the transgenes disrupts endogenous genes, causing artifacts. Some models can only be maintained on specific genetic backgrounds, or produce unexpected results when crossed with other transgenics. Overexpression of APP may lead to ER stress and excessive production of toxic APP fragments other than Aβ, such as β-CTF.
Saido believes his knock-in APPNL-F mice, which carry the Swedish and Iberian mutations, and APPNL-G-Fmice, which add the Arctic mutation, overcome these problems (see Apr 2014 webinar). The knock-ins live a normal lifespan but develop early amyloidosis and inflammation. Memory problems crop up at 18 months in APPNL-F and at six months in APPNL-G-F, which accumulates amyloid faster. A third model, APPNL, carries only the Swedish mutation and has mild, slowly developing pathology.
Several presenters discussed how the mice have performed in their hands. For example, Taisuke Tomita of the University of Tokyo had in vitro evidence that the serine protease kallikrein 7 (KLK7) degrades monomeric and fibrillar Aβ. Tomita generated KLK7 knockout mice, and found they accumulated 50 percent more soluble Aβ40 and Aβ42 than wild-types. Crossing them to APPNL-G-F knock-ins, he saw a sixfold increase in amyloid plaques, 20 percent more tau phosphorylation, and earlier astrogliosis than in the knock-in parents (see image above). The findings establish a role for KLK7 in controlling pathology, Tomita concluded. The knock-in mice provided a good model for this research because they develop robust plaques and neuroinflammation early in life, Tomita told Alzforum. In particular, amyloid deposition varied less among knock-in littermates than it does among overexpression littermates. Because the knock-ins can be maintained as homozygotes, crosses and genome editing are easier to perform, he added. He also found the mice easier to handle. They jump and try to bite their handlers less, and are generally more docile than overexpression models, he said.
Amantha Thathiah, now at the University of Pittsburgh, compared findings from APP/PS1 and APPDutch mice to APPNL and APPNL-F knock-ins. In collaboration with Bart De Strooper at KU Leuven, Belgium, Thathiah had previously reported that in all these models, lowering expression of the G-protein coupled receptor Gpr3, which interacts with γ-secretase, dampened Aβ production (see Oct 2015 news). At SfN, she noted that she saw a more dramatic drop in absolute soluble Aβ levels in the APP/PS1 mice than in knock-ins. This might be expected, since the overexpression model produces more Aβ. On the other hand, in the APP/PS1 mice, decreases in the Aβ42/Aβ40 ratio fell short of statistical significance, whereas in the knock-ins, which have a higher baseline Aβ42/Aβ40 ratio due to the presence of the Iberian mutation, that decrease was clear. “Each model mimics a slightly different aspect of the disease,” Thathiah wrote to Alzforum. “Although the APP knock-in mouse models have late-developing, mild phenotypes that make them less practical for some studies, they avoid problems associated with overexpression and would be useful for testing potential preclinical AD therapies.”
Cognitive Effects Subtle, but Present
At the SfN session, researchers complained most loudly about the lack of a robust behavioral phenotype in the knock-ins. Without behavioral data, papers are more difficult to publish, noted Ilya Bezprozvanny of the University of Texas Southwestern Medical Center, Dallas. Saido said the mice appear to model the early, preclinical stages of AD, when amyloid accumulates and inflammation burgeons, but only subtle cognitive problems are apparent. In people, this stage can last up to 20 years before notable neurodegeneration occurs. The APP knock-in mice, which live only two years, may never reach that later stage.
Nonetheless, the knock-in mice do develop subtle behavioral defects. In a poster, Amira Latif-Hernandez, working with De Strooper and Rudi D’Hooge at KU Leuven, detailed synaptic and memory impairments in these animals. She tested APPNL mice and APPNL-G-F mice at three and six months of age. The APPNL-G-F mice spent more time in open arms of a maze than did the APPNL mice, particularly as they aged, indicating they were less anxious. Learning and memory seemed similar in both models, as they adapted to changes in the location of a hidden platform in the Morris water maze equally well. By six months, however, APPNL-G-F mice froze less often than APPNL mice in a location where they had previously received a shock, suggesting some hippocampal and amygdala deficits, Latif-Hernandez noted.
Corresponding differences cropped up in electrophysiology. Hippocampal slices from six-month-old APPNL-G-F mice showed weaker long-term potentiation than APPNL mouse slices, but similar basal transmission and long-term depression. In slices from the prefrontal cortex, both basal transmission and LTP faltered in the six-month-old APPNL-G-F animals. The mouse scanner revealed increased connectivity of the frontal network in three-month-old APPNL-G-F mice relative to APPNL mice, but no difference at six months. Resting-state functional MRI experiments in older mice are ongoing, Latif-Hernandez reported. “We conclude that an increased Aβ42/Aβ40 ratio in APPNL-G-F mice at six months drives synaptotoxicity, which in turn causes increased deficits in specific behavioral domains,” Latif-Hernandez wrote.
The Best Model? The Answer Is in the Works
How common will use of the knock-ins become? It depends on the lab. Some, such as De Strooper's, are switching over to knock-in models exclusively. Others switch over partially. For example, Bezprozvanny used the knock-ins to investigate disruptions in calcium signaling and plans to do future signaling studies in them, too, though for now he will stick to overexpression models for behavioral data (see Oct 2015 news). Bezprozvanny believes the knock-ins recapitulate Alzheimer’s pathology more faithfully than do overexpression models. He is currently repeating older experiments done with overexpression lines in the knock-ins, but told Alzforum he has not yet found discrepant data.
Conflicting data has turned up in other experiments, however. Saito recently reported two findings from overexpression models that failed to reproduce in the knock-ins: a drop in Nav1.1 sodium channels, and a rise in the CDK5 activator p25 (see Sep 2016 news). At SfN, researchers debated the meaning of the findings, but did not reach consensus. Some argued that the knock-in mice themselves might produce artifacts. Robert Vassar of Northwestern University, Chicago, said that combining several familial mutations is artificial and could cause effects not seen in familial or sporadic AD. The Arctic mutation lies in the Aβ sequence and is known to alter how the peptide aggregates. Lennart Mucke of the Gladstone Institute of Neurological Disease, San Francisco, suggested that overexpression models may represent some aspects of Alzheimer’s disease more faithfully than do the knock-ins. For example, high levels of APP may accumulate at synapses in Alzheimer’s patients, as they do in Tg2576 mice, he said (see Tiwari et al., 2016).
In the end, researchers agreed that whether the new models are better than overexpression transgenics will become clear as more data on the knock-ins come online. “The jury is still out,” David Brody of Washington University, St. Louis, wrote to Alzforum. Mucke summed up the mood when he quoted the statistician George Box: “All models are wrong, but some are useful.” To derive the fullest picture, researchers need to compare several models, with the ultimate validation being human data, Mucke argued.
De Strooper concurred that to be considered truly robust, findings should be consistent across multiple approaches. Overexpression may remain useful for some studies, De Strooper said, especially behavioral ones, but he believes the proper control for those would be mice that overexpress wild-type human protein. Vassar agreed that knock-ins are an additional tool, writing to Alzforum, “The APP knock-in mice are an advance, but all animal models have limitations. We must keep ourselves constantly aware of these limitations and include rigorous controls in our experiments so that we can correctly interpret our results.”
Some researchers believe that adding normal levels of human tau to the mice might improve the model; for more on that, see next SfN story.—Madolyn Bowman Rogers
Next-Generation Mouse Models: Tau Knock-ins and Human Chimeras
Scientists seeking better mouse models for Alzheimer’s disease recently developed human mutant APP knock-ins (see related SfN story). The slowly developing pathology in these mice may better resemble what happens in human brain, where the disease takes decades to manifest, than overexpression models do, noted Takaomi Saido of RIKEN Brain Science Institute, Wako, Japan, who generated the mice. However, for researchers trying to publish quickly, the mild phenotype and scant behavioral defects can be a disadvantage. What’s the answer?
Perhaps tau. APP knock-ins, like most APP overexpression models, lack tau neuropathology and neurodegeneration, defining aspects of human AD. The mice appear to model early, amyloid-only stages of AD. Is this due to the mice’s lifespans, or could it be due to differences between human and mouse tau? At a symposium at the Society for Neuroscience annual meeting, held November 12-16 in San Diego, Takashi Saito from Saido’s group debuted a mouse with wild-type human tau knocked in. In preliminary data, crossing this mouse with the APP knock-ins resulted in hyperphosphorylated, insoluble tau and neuronal death, Saito reported. He emphasized that the hTau knock-ins are available to academic and industry scientists who use the APP knock-in models under the same material transfer agreement. The Japanese group asked those interested to contact saido@brain.riken.jp. Another researcher at the symposium presented a cross of the APP knock-ins with transgenic mice overexpressing human tau; this combination boosted hippocampal atrophy. Such crosses could better model later stages of AD, researchers suggested.
Meanwhile, Bart De Strooper of KU Leuven, Belgium, described another approach to simulate Alzheimer’s disease. Because knock-in mice still have mouse neurons, which behave differently than human cells, his group added human induced pluripotent stem cells to transgenic mouse brain to study how amyloid pathology affects human neurons in the context of a multicellular brain environment (see below).
Trigger Neuronal Death.
Neurons (nuclei blue) in the entorhinal cortex of 18-month-old hTau/APPNL-F mice activate caspase-3 (red), indicating apoptosis. [Courtesy of Takaomi Saido and Takashi Saito.]
How Does Tau Fit In?
Researchers at the symposium complained about how slow research with the knock-in models is. Would adding pathological tau jack up the phenotypes? Jennifer Macdonald, working with Michel Goedert at MRC Laboratory of Molecular Biology, Cambridge, England, crossed heterozygous P301S tau mice with APPNL-F mice. At 18 months of age, the offspring had lost more hippocampal volume than their P301S parents. APPNL-F mice, by contrast, maintain normal-sized hippocampi even at two years old. The data suggest a synergistic effect of Aβ and tau on neurodegeneration.
Aβ and tau pathology converged in the subiculum of the P301S/ APPNL-F mice. There, Macdonald saw dramatic changes in microglial cells. In wild-type mice, most microglia have the normal ramified shape and only 13 percent are dystrophic with degenerating processes, whereas in P301S and APPNL-F parent mice, one-third are dystrophic. In the APPNL-F/P301S crosses, more than half the microglia were dystrophic. Rod-shaped microglia, which are associated with neurological infections and traumatic brain injury (see Bachstetter et al., 2015), make up 2 percent of the total in APPNL-F mice, 6 percent in P301S mice, and almost 10 percent in the APPNL-F/P301S crosses. Furthermore, the crosses had twice as many activated microglia, as seen by Iba1 staining, as their parents did.
The pronounced increase in dystrophic and rod-shaped microglia is similar to what is seen in AD brain, Macdonald noted. “The cross recapitulates at least two main features seen in AD: hippocampal atrophy, and microglial changes,” she wrote to Alzforum.
The overexpression problem applies to tau transgenic mice, too. The P301S mice overexpress human mutant tau, but most cases of Alzheimer’s involve normal levels of wild-type tau. To investigate the effects of normal human tau, Saito generated tau knock-in mice by humanizing the mouse tau gene. People express six tau isoforms, whereas wild-type mice express only three. The knock-in mice produce all six, Saito reported in San Diego. The animals appeared healthy and made normal amounts of tau that stayed in its customary axonal location.
Saito then crossed the hTau knock-ins with APPNL-F mice. The offspring accumulated more phosphorylated and insoluble tau than their hTau knock-in parents. Compared to the APPNL-F knock-ins, inflammatory cytokines and activated microglia were similar, but more neurons were dying by 18 months. Regions of neuronal death did not correlate with the pattern of amyloid deposition, as is also the case in AD. The hTau/APPNL-F mice activated more caspase-3 in the entorhinal cortex at 18 months (see image above). Notably, however, no neurofibrillary tangles had appeared by this age. Analysis is ongoing, with some preliminary data suggesting the entorhinal cortices of these crosses start to shrink at 21 months. The fact that the entorhinal cortex degenerates in the absence of tangles hints that oligomeric tau might be the culprit, Saido speculated.
A Chimeric Approach: Human Neurons in Mouse Brain
While the knock-in mice with humanized Aβ and tau may more faithfully model AD, they still do so with mouse neurons. Do those behave differently than human ones? De Strooper took a radically different stab at modeling AD, which allows him to ask how human neurons are affected by amyloid pathology in the complex environment of the brain. He implanted human induced pluripotent stem cells from healthy controls into the brains of newborn APP/PS1 mice, and examined brain pathology two, four, six, and eight months later.
In the 165 mice investigated to date, De Strooper and colleagues have found consistent results. Human-derived grafts develop similar levels of inflammation as surrounding mouse brain tissue, but slightly fewer amyloid plaques. Tau in the grafts becomes hyperphosphorylated, but does not form neurofibrillary tangles. However, grafts display more presynaptic pathology than host tissue, and massive neuronal death. De Strooper noted that the cell death appears necrotic, rather than apoptotic. When human cells are transplanted into wild-type mice, or mouse neurons are transplanted into the APP/PS1s, the transplanted neurons stay alive. The results suggest a unique vulnerability of human neurons to amyloid pathology, which does not depend on the presence of tau tangles, De Strooper noted. He is now analyzing gene expression changes in the grafted neurons and surrounding cells to glean clues as to why this happens.—Madolyn Bowman Rogers
Inflammation Helps Microglia Clear Amyloid from AD Brains
Neuroinflammation can both help and harm the Alzheimer’s brain, leaving researchers uncertain of the best way to modulate the immune system therapeutically. Previously, many scientists thought that dialing down inflammation would help, but growing evidence now suggests the opposite. Numerous talks at the annual Society for Neuroscience conference, held November 12-16 in San Diego, reinforced this new view, with converging lines of evidence for the benefits of a pro-inflammatory brain milieu. Specifically, pumping up the appetites of microglia clears amyloid and improves cognition in mouse models, researchers said. They put forward several different ways to stimulate microglia, including cytokines and an antibody that recognizes the microglial receptor TREM2. Other presentations drilled into how aggregated Aβ and TREM2 promote a pro-inflammatory state. Overall, the data suggest an emerging consensus on how inflammation could be harnessed to slow AD.
“Over the last decade, we have been trying to advance the hypothesis that the inflammatory response in AD is ineffective because it is out of balance. Paradoxically, it isn’t too much inflammation that seems to be the culprit in AD; it’s not having the right kind of immune response,” Terrence Town of the University of Southern California, Los Angeles, wrote to Alzforum. In particular, he believes TREM2 expression might be suboptimal in the AD brain, and that stimulating TREM2 signaling might be one way to restore balance.
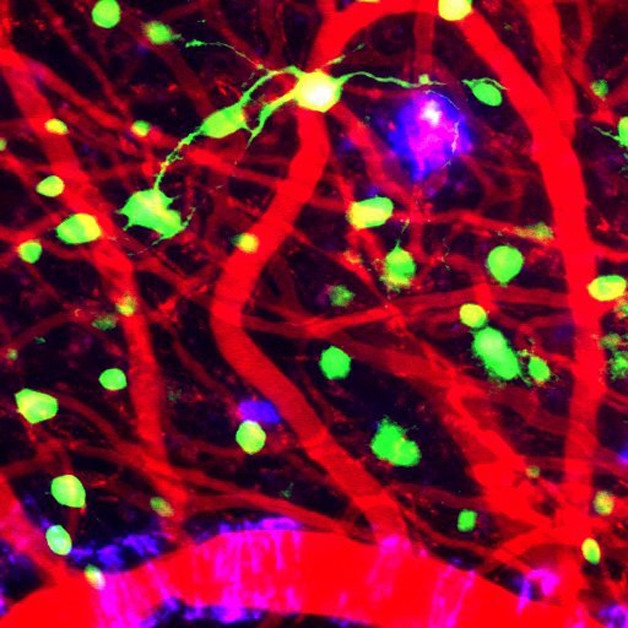
Too Little TREM2?
In an AD brain section, activated microglia (red) surround an amyloid plaque (green), but express sparse TREM2 (pink). [Courtesy of Brian Leung and Terrence Town.]
Some previous studies had already cast inflammation as a force for good in Alzheimer’s. Todd Golde of University of Florida, Gainesville, reported that the pro-inflammatory cytokine IL-6 stimulates gliosis, enhancing Aβ phagocytosis in transgenic mice (see Chakrabarty et al., 2010). Both Golde and Town separately found that the anti-inflammatory cytokine IL-10 worsens amyloid pathology and cognition in mice (see Feb 2015 conference news). Not all studies agree, however, with other researchers reporting negative effects from different pro-inflammatory cytokines (see Nov 2012 news).
For his current research, Golde and colleague Yona Levites wondered how the inflammatory state of the brain might affect AD immunotherapy. To address this, Levites injected a vector carrying either IL-6 or IL-10 into the brains of newborn CRND8 mice, which overexpress mutant human APP and typically develop amyloid plaques by three months of age. Beginning two months after birth, the researchers injected 0.5 mg of the in-house anti-Aβ1-16 antibody mAb5 intraperitoneally into half the transfected mice (see Levites et al., 2015). The animals received injections twice weekly for four months.
When the animals were analyzed at six months, mice that expressed exogenous IL-6 had less amyloid than untreated controls. Antibody treatment on top of that lessened the load a little further, displaying a partial additive effect with IL-6. In mice that received both IL-6 and mAb5, insoluble Aβ42 and Aβ40 levels dropped nearly in half, while SDS-soluble Aβ crashed by two-thirds. On the other hand, mice expressing IL-10 fared worse than untreated controls, developing a heavier amyloid load. Moreover, the presence of IL-10 blunted the benefits of antibody treatment, Golde reported.
The data suggest that the brain’s underlying immune status can modulate the outcome of immunotherapy, Golde said. He noted that immune activation follows the “Goldilocks Principle”—it needs to be just right. Recent mouse data point to benefits from turning up the immune system to run a little hotter in the AD brain, Golde said. An audience member wondered if the findings will translate to people, since both IL-6 and IL-10 have been reported to be elevated in AD brain. However, Golde noted that most of those data come from postmortem brains. People with AD often die from infections such as sepsis or pneumonia, which could alter the immune status of the brain. Researchers need to assess the level of innate immune factors in living patients, he suggested. If the data show that immunotherapy does require pro-inflammatory factors to be most effective, researchers might borrow a technique from the cancer field and create “chemobodies” that combine an antibody with another molecule to accomplish both goals at once, Golde suggested.
Others were not so sure. Gary Landreth of Case Western Reserve University, Cleveland, noted that overexpression of IL-6 and IL-10 in these experiments makes it hard to determine how the findings apply to normal physiology, or what this means for phagocytosis. “There is little mechanistic understanding of how a pro- or anti-inflammatory milieu interacts with the phagocytic machinery in the brain,” he wrote to Alzforum.
What else besides cytokines affects microglial activation? Several presentations fingered the microglial receptor TREM2, variations in which triple the risk of AD. Many researchers now believe this is because these variants hamper the ability of microglia to contain or clear plaques (see Nov 2012 news; May 2016 news; Jul 2016 news).
At SfN, Taylor Jay and Margaret Broihier at Case Western further elucidated how TREM2 affects microglia. Working with Landreth and Bruce Lamb at Indiana University, Indianapolis, Broihier compared APPPS1 mice with normal levels of TREM2 to mice that lack the protein. Microglia from the TREM2 knockouts expressed lower levels of several pro-inflammatory markers, such as IL-1β, IL-6, iNOS, TLR4, and TNFβ, and higher levels of anti-inflammatory markers such as Fizz1, Arg1, and TGFβ, supporting the idea that TREM2 promotes inflammation. TREM2 knockout microglia proliferated poorly and tended to die more readily than those with the receptor. That said, the presence or absence of TREM2 did not affect the cells’ ability to gobble up fluorescent beads, contradicting some previous studies, Broihier reported (see Jul 2014 webinar).
If TREM2 helps trigger beneficial inflammation, could it be targeted therapeutically? Jennifer Gooch at the University of Kentucky, Lexington, working with Donna Wilcock there, investigated this question. She used a commercial anti-TREM2 antibody made by the San Francisco-based biotech company Alector that activated signaling through TREM2 and its co-receptor, DAP12. The antibody did not bind to the brains of TREM2 knockout mice, confirming its specificity. Gooch injected the antibody into the bilateral frontal cortices and hippocampi of APPPS1 mice. After 72 hours, she sacrificed the mice and examined their microglia. Antibody treatment had boosted the amount of pro-inflammatory, healing, and repair markers expressed by microglia, while roughly doubling immunoreactivity of CD11b, a marker for microglia. Amyloid deposits in the vicinity of the injection, as seen by immunohistochemistry, had fallen by one-third to one-half compared with mice that received a control antibody. This is comparable to the reduction in total Aβ seen after injecting an anti-Aβ antibody in the same regions, Wilcock noted (see Wilcock et al., 2003). Activating TREM2 with an antibody appears to stimulate the phagocytosis of Aβ, and might make a promising therapeutic, Gooch suggested. The researchers next plan to dose mice systemically with the anti-TREM2 antibody once weekly for several weeks, and examine the effects on behavior. “Our data indicate that TREM2 activation promotes amyloid clearance, but whether this results in a functional benefit remains to be determined,” Wilcock wrote to Alzforum.
Other speakers proposed different ways to stimulate microglia. Alex Vesling, in Town’s group at the University of Southern California, Los Angeles, turned to the toll-like receptor (TLR) signaling pathway. Aggregated Aβ binds TLRs, activating signaling through IL-1 receptor-associated kinases (IRAKs) and TNF-receptor associated factor 6 (TRAF6). This pathway switches on NF-κB and cranks up inflammation (see Sep 2009 news). The response can be inhibited by activation of IRAK-M, a protein found only in microglia and macrophages. Thus, IRAK-M dampens inflammation in microglia. Notably, IRAK-M has been reported to be dysregulated in aging and AD (see Cribbs et al., 2012).
Vesling examined hippocampal lysates from postmortem AD brains, and found an excess of cleaved, non-functional IRAK-M, as well as an abundance of TRAF6, suggestive of increased inflammation. Vesling then transfected human microglial cultures with IRAK-MΔDD, a mutant form that mimics the cleaved product, before treating the cells for two or six hours with aggregated Aβ42. TRAF6 preferentially binds IRAK-MΔDD over the wild-type IRAK-M, suggesting the mutant should have a dominant-negative effect, Vesling noted. Indeed, in cells that expressed IRAK-MΔDD, TRAF6 expression rose, along with Aβ uptake and expression of pro-inflammatory cytokines including IL-6 and TNFα. Expression of anti-inflammatory cytokines such as TGFβ dropped. Inhibiting IRAK-M pumps up phagocytosis, Vesling concluded. He speculated that cleavage of IRAK-M in AD brain is a compensatory response to try to clear amyloid, but perhaps it occurs too late to help. Activation of TRAF6 may represent a pharmacologic target, he suggested.—Madolyn Bowman Rogers
Exosomes and Antibodies Tie Plasma Aβ and Tau to Alzheimer’s
This story was updated on 16 December 2016 to add a paragraph about exosomes in Down’s syndrome.
While changes in cerebrospinal fluid Aβ and tau reliably flag the onset of Alzheimer’s disease, plasma levels of these proteins have remained frustratingly uninformative. At the annual Society for Neuroscience conference, held November 12-16 in San Diego, speakers once again wrestled with the question of what these plasma proteins reflect, and how to detect the forms most related to pathology.
Researchers in Indiana reported that CSF and plasma tau, both of which rise in AD, each correlated with atrophy of a different set of brain regions. Meanwhile, other speakers presented approaches that home in on particular species of plasma Aβ and tau, such as oligomeric forms and those packaged in exosomes coming from the central nervous system. The neurosteroid allopregnanolone also made a showing as a potential prognostic marker. While preliminary results indicate that these various markers can pick out people on the path to AD in small samples, it remains to be seen whether any of them will prove robust.
“Progress on blood biomarkers is encouraging,” Andrew Saykin at the Indiana University School of Medicine, Indianapolis, wrote to Alzforum. “Plasma tau seems to be one of the most promising ones. The work on neuronal exosomes from peripheral blood is also exciting, and I am glad to see reports of replication.”
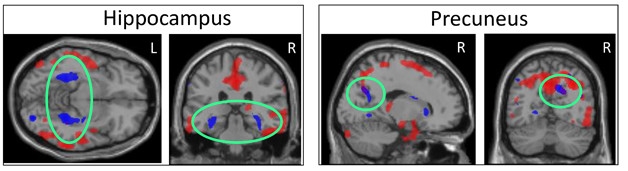
Blood and CSF Tell Different Tales. High plasma tau correlates with atrophy in specific subcortical regions (blue), while high CSF tau relates to shrinkage of largely cortical areas (red). The two patterns overlap only in precuneus (right, green circles). [Courtesy of Kacie Deters and Andrew Saykin.]
The quest for plasma biomarkers has been littered with disappointments, with many initially promising candidates failing to replicate (see Jun 2013 webinar; Feb 2016 news). A recent meta-analysis found that among the most commonly investigated plasma biomarkers, only total tau levels distinguished AD patients from controls (see Apr 2016 news). Plasma tau runs about twice as high in AD patients, but only weakly correlates with CSF total tau, leaving it unclear what these high levels signify (see Mattsson et al., 2016). One problem is that plasma tau is so low that it is difficult to measure accurately. The recent development of ultrasensitive single-molecule assays (Simoa) now allows researchers to detect quantities in the low pg/ml range or below, making measurements more reliable and opening up new frontiers for plasma biomarkers (see Apr 2016 conference news).
To find out how plasma tau relates to neurodegeneration, Kacie Deters, working with Saykin at Indiana, correlated plasma and CSF tau with brain atrophy as seen on structural MRI scans. She used data from 331 participants in ADNI-1, about one-quarter of whom were cognitively normal, half of whom had symptoms of mild cognitive impairment, and the remainder a clinical diagnosis of AD. Plasma total tau was measured with Simoa.
Deters found that a person’s plasma total tau concentration correlated largely with their degree of shrinkage in subcortical brain regions, including the parahippocampus, hippocampus, and striatum, with only a little cortical involvement in the precuneus. CSF total tau, on the other hand, correlated best with atrophy of cortical regions, particularly the medial temporal cortex. The atrophy patterns overlapped in a few brain regions, such as the precuneus, but were largely separate. Peripheral and central measures of tau may reflect different aspects of AD, or involve different tau isoforms, Deters speculated. In other studies, high plasma tau has been linked to recent brain injury and subcortical axon damage (see Mar 2014 news). Deters suggested that plasma tau could be used to screen people to find out who might need additional tests and imaging.
Other researchers are focusing on specific forms of plasma tau or Aβ. Dimitrios Kapogiannis at the National Institute on Aging, Baltimore, had previously reported that exosomes that enter blood from the central nervous system can be used to identify people with AD. Specifically, neuronally derived plasma exosomes in 57 AD patients carried more Aβ42, p-tau181, and p-tau396 than those from 57 controls, and these proteins were already elevated as early as 10 years before symptoms developed (see Aug 2014 conference news; Fiandaca et al., 2014). Because the findings came from a small cohort, the researchers are now attempting to replicate them in the Baltimore Longitudinal Study of Aging.
At SfN, Maja Mustapic in Kapogiannis’ group showed preliminary results from this cohort. She analyzed plasma samples from 350 participants, 128 of whom developed AD over the course of the study. Each participant had three samples available for analysis, spanning a four-year time period up to their diagnosis of Alzheimer’s. In neuronal exosomes isolated from the AD patients, p-tau181 and p-tau231 were higher than in controls at all time points, Mustapic reported. In contrast, total tau remained similar. The findings were highly significant and matched the data from previous studies, Kapogiannis told Alzforum. Mustapic is still analyzing Aβ42 and other markers in the exosomes. Next steps include combining Aβ and tau to develop a test that better discriminates between patients and controls, she told the audience.
Others described similar findings. In a poster, Charisse Winston, working with Robert Rissman at the University of California, San Diego, reported that exosomes from 10 AD patients and 20 people with mild cognitive impairment who progressed to AD contained more Aβ42, p-tau181, and p-tau396 than exosomes from 10 controls and 20 people with stable MCI. In addition, exosomes from people on the path to AD carried less of the synaptic protein neurogranin and the protective transcription factor REST than did those from controls (see Mar 2014 news). In particular, high p-tau181 and low neurogranin predicted progression to AD over the three years of the study, she noted. The lab will next investigate how this combination of five markers changes over time, and whether it can be used to stage AD.
In a separate experiment, the researchers injected these human exosomes into the hippocampi of young wild-type mice. One month later, levels of p-tau and aggregated tau had mushroomed in mice that received exosomes from AD patients. Exosomes from MCI patients seeded milder pathology, and those from controls, none (see Winston et al., 2016). This suggested that exosomes carry more aggregated tau as disease advances.
Plasma exosomes might also help detect AD in people with Down’s syndrome, suggested Eric Hamlett of the Medical University of South Carolina (MUSC), Charleston. Hamlett and colleagues quantified Aβ42 and p-tau in neuronally derived plasma exosomes from people with Down’s syndrome and controls across a range of ages. At SfN, Hamlett reported that neuronal exosomes from people with DS carried two to three times more Aβ42 and p-tau than those from controls, even early in life. While these markers did not correlate with cognitive status, levels of p-tau396 did. This marker shot up in people with DS who had symptoms of dementia (see Hamlett et al., 2016).
Antibodies offer a different approach to finding pathological forms of Aβ and tau in blood. Michael Sierks at Arizona State University, Tempe, previously generated several single-chain antibody fragments that identify oligomeric forms of Aβ and tau, as well as pathogenic forms of TDP-43 (see Apr 2011 conference news; Kasturirangan et al., 2010; Kasturirangan et al., 2013). These antibodies can capture femtomolar quantities of oligomers from blood and postmortem human brain samples, the researchers report (see Williams et al., 2015).
At SfN, Stephanie Williams in Sierks’ group presented preliminary results from a tiny longitudinal study of four AD patients and two age-matched controls. Their plasma samples spanned the time frame from before they developed symptoms to after their diagnoses. Before symptoms, plasma samples from the AD-bound volunteers harbored a low concentration of tau oligomers, but more pathological Aβ and TDP-43 than controls. After a diagnosis of mild cognitive impairment, tau and Aβ oligomers rose, while toxic TDP-43 fell. At the dementia stage, Aβ and tau oligomers were both high, while TDP-43 was low, Williams reported. Differences from controls were first apparent seven years before MCI developed, suggesting these markers could predict decline, she noted. Supporting this, higher levels of Aβ oligomers in the blood correlated with faster cognitive decline. The researchers are now repeating the study in a larger cohort.
What about other plasma markers? Junming Wang of University of Mississippi Medical Center, Jackson, investigated whether allopregnanolone, a metabolite of the hormone progesterone, could make a good biomarker. This hormone is in scant supply in the prefrontal and temporal cortices of Alzheimer’s patients, and its lack correlates with how severe pathology has become (see Marx et al., 2006; Naylor et al., 2010). Low allopregnanolone has been reported in the plasma of AD and Parkinson’s patients (see Smith et al., 2006; di Michele et al., 2003). Allopregnanolone is currently in a Phase 1 Alzheimer’s trial led by Roberta Brinton at the University of Southern California (see Aug 2013 conference news; Dec 2014 conference news). Wang previously worked with Brinton, but she is not involved in his current research.
To see if allopregnanolone could be a marker in AD, Wang is analyzing a cohort of 16,000 people followed at UMiss for 30 years. They donated plasma samples five times during the course of the study, starting at the beginning when everyone was still cognitively healthy. Wang and colleagues are using Simoa to measure allopregnanolone quantities down to fg/ml, he said. Preliminary data suggest this method can detect small decreases in the hormone about 10 to 15 years before cognitive symptoms appear, hinting it might be prognostic. Wang is still analyzing how well these low allopregnanolone levels predict future risk of neurodegenerative disease.—Madolyn Bowman Rogers
Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, Palmqvist S, Baker D, Tan Hehir CA, Jeromin A, Hanlon D, Song L, Shaw LM, Trojanowski JQ, Weiner MW, Hansson O, Blennow K, ADNI Investigators.
Plasma tau in Alzheimer disease.
Neurology. 2016 Oct 25;87(17):1827-1835. Epub 2016 Sep 30
PubMed.
From Stem Cell Exosomes to Restoring ZZZs: New Ideas to Protect the Brain
The dearth of new drugs to treat neurodegenerative disease is making researchers cast their nets wide as they explore new ideas for prevention and treatment. Some creativity was on display at the annual Society for Neuroscience conference, held November 12-16 in San Diego. There, speakers proposed restoring sleep patterns to prevent amyloid plaque accumulation, and regulating an inflammatory astrocyte pathway to ward off vascular dementia. Studying the resilience of some elders’ brains to Alzheimer’s pathology, other scientists unearthed a new explanation: These brains generate high numbers of neural stem cells that release exosomes packed with a suite of regulatory microRNAs that preserve synapses. Most of these ideas have far to go before they could become usable therapies, but the approaches sparked excitement at SfN.
Tracking Amyloid Accumulation. A cranial window allows researchers to follow the development of plaques (purple), and their relationship to neurons (green) and blood vessels (red), in the cortices of live AD model mice after sleep therapy. [Courtesy of Brian Bacskai and Ksenia Kastanenka.]
New Clues to Cognitive Reserve
Researchers have long been tantalized by the observation that some people resist the deleterious effects of Alzheimer’s pathology into very old age (see Oct 2016 news). For example, Changiz Geula of Northwestern University, Chicago, previously reported that the brains of some old people with superior cognitive ability harbored extensive amyloid plaques (see Dec 2008 conference news). At an SfN press conference on AD, Geula extended this with a detailed look at eight people who had died in their late 90s, having maintained the recall prowess of a 50- or 60-year-old. Postmortem, their brains revealed a wide range of pathology, he said. Two had limited plaques and tangles equivalent to Braak stage I or II, four were classified as Braak stage III or IV, and the remaining two brains contained enough plaques and tangles to meet a pathological diagnosis of Alzheimer’s disease. However, all these brains contained healthy-looking neurons, not the extensive cell death seen in people with clinical symptoms of AD. In addition, brains of similar cases sported more postsynaptic markers than the brains of age-matched controls with average cognitive abilities. The data suggest that these people possess resilience factors that protect their brains, Geula concluded, and he is trying to identify those from among a range of genetic and environmental parameters.
In a separate talk, Maria Micci, who works in collaboration with Giulio Taglialatela at the University of Texas Medical Branch, Galveston, proposed an explanation for why some older people maintain their synapses so well. The researchers had noticed that the brains of people who died with preserved cognition but extensive Alzheimer’s pathology contained more neural stem cells (NSCs) than did AD or MCI brains. The higher the number of these cells, the better the person’s score had been on their last MMSE (see Briley et al., 2016). When they examined the brains more closely to see why cognition might be spared, Taglialatela and colleagues found that postsynapses remained devoid of Aβ oligomers. This contrasted with the high levels of synaptic Aβ in AD brains, as seen by antibody staining of isolated synapses (see Bjorklund et al., 2012).
How might the NSCs protect synapses? Perhaps, Micci thought, via exosomes. After all, these vesicles are packed with cargo and can be taken up by bystander cells. To test this, she treated mouse hippocampal slice cultures with exosomes secreted by cultured rat NSCs, then later added preformed oligomers of recombinant Aβ42. Pretreatment with NSC exosomes spared these hippocampal synapses from accumulating Aβ, she reported. The treatment also preserved long-term potentiation and prevented synaptic excitability.
Micci saw similar results when she injected rat NSC exosomes into the hippocampi of wild-type mice, waited four hours, then isolated synaptosomes and treated them with Aβ in vitro. Again, synapses stayed clean. Next, the researchers examined nestin-δ-HSV TK-eGFP mice, in which neurogenesis can be switched off. After disabling neurogenesis and isolating synapses, Aβ treatment resulted in excessive oligomers crowding these structures. As in wild-type mice, this experimental inundation of synapses was prevented by injecting NSC exosomes into the brain before isolating synapses. In other experiments, the researchers have found that NSC exosomes shield synapses from tau oligomers as well, Taglialatela told Alzforum.
Notably, exosomes from mature neurons conferred no protection in any of these scenarios. To find out what makes NSC exosomes special, the researchers broke them open and compared their contents to those of neuronal exosomes. That revealed a unique signature of eight miRNAs in the NSC exosomes. Now, Micci and Taglialatela are exploring whether this set of miRNAs affects how synapses function, and whether it can protect them from damaging oligomers. Micci will also isolate exosomes from postmortem AD and control brains to see if they contain the same set of miRNAs.
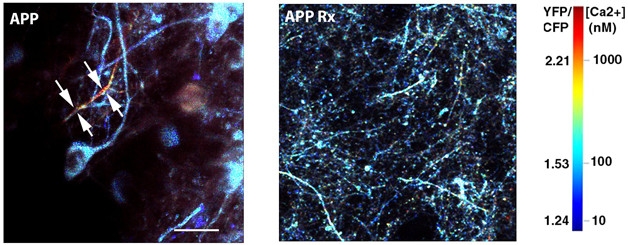
Calming Calcium Spikes. In AD model mice (left), high calcium levels (yellow and red) flood neurons (blue), but disappear when slow-wave sleep is restored (right). [Courtesy of Brian Bacskai and Ksenia Kastanenka.]
Can Refreshing Circadian Rhythms and Sleep Block Plaque Accumulation?
Researchers have long known that AD patients sleep poorly, but recent studies have switched things around to suggest disrupted sleep may precede and contribute to the development of dementia (see Aug 2012 conference news; Oct 2013 news). In part, this may be because the brain clears waste proteins such as amyloid during slumber (see Jun 2014 news; May 2014 conference news). Speakers at SfN suggested two different approaches to normalizing sleep, with an eye to preventing amyloid buildup.
Trongha Xuan Phan, who works with Li-Huei Tsai at the Picower Institute at Massachusetts Institute of Technology, Cambridge, and Robert Vassar at Northwestern University, Chicago, focused on the circadian clock and the genes it regulates. Some of those might be therapeutic targets for restoring Aβ clearance, he reasoned. Because histone deacetylase 1 (HDAC1) knockout mice have abnormal circadian rhythms, he investigated which genes this enzyme controls. In knockout mice, preliminary results suggest that only 1,421 genes oscillated with the sleep-wake cycle, compared to 2,822 genes in wild-type mice, he reported. Thus, HDAC1 appears to control about half of normally cycling genes. Loss of cycling correlated with worse contextual memory in these mice; this dovetails with other studies tying circadian disruptions to poor hippocampal function (see Wardlaw et al., 2014).
Among the genes with disrupted cycling, aquaporin 4 stood out to the researchers. This astrocytic water channel has been associated with clearance of Aβ from the brain, and has been found to be in short supply in AD brains (see Aug 2012 news). Surprisingly, Phan found that the HDAC1 knockouts expressed twice as much aquaporin 4 as controls, rather than less. Looking more closely at previous research for an explanation, he found that the site of aquaporin 4 expression matters. Global brain expression has been reported to be elevated in AD patients, even while aquaporin 4 near blood vessels drops (see Hoshi et al., 2012; Dec 2016 news). Since Phan measured global aquaporin 4 expression in the hippocampus, the findings are consistent with the literature, he told Alzforum.
The researchers have now crossed HDAC1 knockout mice with an AD model to find out if loss of circadian rhythms affects pathology. In the future, Phan plans to examine how aquaporin 4 expression in the perivasculature changes during the day. Global aquaporin 4 expression peaks during sleep in people, but these oscillations dampen with age, Phan noted. He also wants to study how light therapy, which helps improve sleep in older adults and people with AD, affects HDAC1, circadian gene cycling, and Aβ clearance.
Ksenia Kastanenka, working with Brian Bacskai of Massachusetts General Hospital, Charlestown, took a different tack. Recent studies have suggested that amyloid accumulation in the brain can disrupt slow-wave sleep, which is essential for memory consolidation (see Jun 2015 news; Varga et al., 2016). Kastanenka used mice to take a closer look at this issue. To visualize slow-wave oscillations, she applied a voltage-sensitive dye to the cortices of APPswe/PS1DE9 mice through a cranial window. She found that the power, but not the frequency, of the oscillations dropped starting at three months of age, before plaques formed.
What might explain this? One clue was that these mice have low levels of the inhibitory neurotransmitter GABA. When the researchers injected exogenous GABA into their brains, it restored the power of slow-wave sleep, suggesting that overactive neurons were responsible for the disruptions. Other studies agree that neuronal hyperactivity contributes to memory problems and degeneration in AD (see Dec 2011 news; Mar 2015 news).
To see if Aβ by itself could disrupt slow waves, Kastanenka and colleagues collected Aβ oligomers secreted by cultured neurons isolated from Tg2576 mice. They added these oligomers to the cortices of wild-type mice through a cranial window. Again, they saw a drop in the power of slow-wave oscillations.
Could restoring slow-wave sleep help neurons? The researchers expressed light-activated channelrhodopsin 2 in frontal cortical neurons of three-month-old APPswe/PS1DE9 mice, waited one month, then drove neuronal activity with blue light delivered to the cortex. When light flickered at the frequency of slow waves, the power of these oscillations returned to normal. The treatment boosted GABA to wild-type levels and dampened neuronal calcium, another marker of hyperactivity, to normal levels (see image above).
Moreover, during two months of light treatment, no new amyloid plaques formed. These mice normally deposit numerous plaques at that age. Altogether, the data suggest that restoring slow-wave sleep in AD patients might prevent neurodegeneration, Kastanenka said. An enthusiastic audience peppered her with questions, and wanted to know how these findings might be applied. Kastanenka suggested that the key might be to enhance the activity of inhibitory interneurons to restore balance to the circuitry in AD brains.
Targeting Astrocytes to Ameliorate Vascular Dementia
Could astrocytes be the ticket to new therapies beyond AD? Christopher Norris of the University of Kentucky, Lexington, thinks as much. He focuses on vascular dementia, the second leading cause of dementia that co-occurs in almost half of AD cases. Despite numerous hypotheses, researchers have reached no consensus for how vascular problems cause cognitive decline. Because astrocytes act as liaisons between blood vessels and neurons, Norris wondered if they might play a role. To investigate, he used a mouse model of vascular dementia developed by colleague Donna Wilcock at Kentucky. Wild-type mice are fed a diet rich in methionine and low in folate, boosting levels of homocysteine in the blood, damaging blood vessels, and causing cognitive decline (see Sudduth et al., 2013; Sudduth et al., 2014). In these mice, the astrocyte end-feet that wrap around blood vessels become disrupted, losing crucial ion channels, Wilcock found (see Sudduth et al., 2016).
What might explain these changes? In AD models, Norris and others previously reported that Aβ stimulates calcium release in astrocytes, thus activating the phosphatase calcineurin, which in turn triggers the transcription factor NFAT. This pathway turns on inflammation and triggers loss of synapses and cognitive decline (see Oct 2009 news; Dec 2009 news; Feb 2010 news). Norris thought this pathway might be activated by vascular pathology, as well. After all, astrocytes near microinfarcts in postmortem brain sections express high levels of activated calcineurin (see Pleiss et al., 2016).
If so, could turning down NFAT signaling ameliorate vascular dementia? Norris and colleagues injected a viral vector carrying the NFAT inhibitor VIVIT into the brains of two-month-old wild-type mice. An astrocyte-specific promoter limited expression to these cells. Two months later, the researchers placed the mice on the vascular dementia diet for three months. Mice whose NFAT was blocked preserved normal long-term potentiation and synapse number around the injection area, whereas controls had deficits on these readouts. In addition, the treated brain region maintained normal cerebral blood flow and healthy neurons. Norris is now testing behavior in treated mice. He suggested that NFAT inhibition might be a viable strategy for preventing vascular dementia. In previous work, he reported that the same approach can rescue synapses and cognition in animals models of AD and traumatic brain injury (see Furman et al., 2012; Furman et al., 2016).—Madolyn Bowman Rogers
Large Phase 2 Trial Starting Up in Genetic Parkinson’s Population
In recent years, a growing number of companies have started to look for ways to stem the progression of Parkinson’s disease. One approach targets glucocerebrosidase (GBA), the leading genetic risk factor for sporadic PD and dementia with Lewy bodies (DLB). Defects in this lysosomal enzyme cause a buildup of α-synuclein, as well as of the glycolipid glucosylceramide that GBA normally cleaves (see Jun 2011 news; Jul 2011 news). Parkinson’s patients with GBA mutations, who make up about 7 percent of cases, decline faster and have more severe symptoms than idiopathic PD patients.
At the annual Society for Neuroscience conference, held November 12-16 in San Diego, Pablo Sardi of Sanofi Genzyme, Framingham, Massachusetts, announced the launch of a Phase 2 study in Parkinson’s patients who carry GBA mutations. The company will test its small-molecule inhibitor of glucosylceramide synthesis, GZ/SAR402671, which turns off production of this key GBA substrate. Because glucosylceramide stabilizes α-synuclein oligomers, its buildup is believed to contribute to α-synuclein toxicity. Also known as ibiglustat, GZ/SAR402671 can be taken by mouth and enters the brain well, Sardi noted. The Sanofi trial will be the largest attempt so far to target treatment to this genetically defined Parkinson’s population. “I think of this as precision medicine for Parkinson’s,” Sardi wrote to Alzforum.
At SfN, Sardi detailed preclinical findings with the closely related compound GZ667161. The researchers fed this compound to PD model mice, which carry the homozygous GBA mutation D409V, from the ages of six to 13 months. Compared with untreated, age-matched controls, GZ667161 slashed hippocampal α-synuclein deposits in the aged mice to less than half, below the amount present when treatment began at six months. Memory in a contextual fear-conditioning test improved. While untreated mice froze only 5 percent of the time in a location where they had previously received a shock, treated animals froze 30 percent of the time, nearing the wild-type recall of 50 percent. Sardi expects to publish these findings soon.
A Phase 2 trial has been approved by the Food and Drug Administration and will begin enrolling this month, with completion scheduled for 2022, Sardi said. The study will enroll about 230 people who have Parkinson’s and a GBA mutation. Although this initial trial only includes mutation carriers, the researchers believe the strategy may work to rein in α-synuclein in other forms of PD as well. Supporting this idea, GZ667161 also suppressed pathology and boosted cognition in the PrP-A53T-SNCA mouse model, which overexpresses α-synuclein but has wild-type GBA, and did so with a similar efficacy to that seen in the D409V mice, Sardi reported.
Michael Schlossmacher at Ottawa Hospital Research Institute, Ontario, Canada, who collaborated with Sardi on the preclinical studies of GZ667161, told Alzforum he is cautiously optimistic that the strategy might help sporadic PD patients, as well as people with DLB. As many as 25 percent of the latter carry GBA mutations, and they typically progress faster than PD patients, so their need for treatment is great, Schlossmacher noted. Sardi said his company will wait to analyze results from the PD genetic cohort before considering whether to test the compound in other groups.
GZ/SAR402671 is currently in a Phase 2 trial for the lysosomal storage disorder Gaucher’s disease, as well as Phase 2 for the related disorder Fabry’s disease. It has been fast-tracked by the Food and Drug Administration for the latter disorder.
Schlossmacher noted that these Gaucher studies provided proof of principle that GZ/SAR402671 can lower accumulation of glucosylceramide in peripheral organs.
Meanwhile, many other companies, including Pfizer and the biotech company Lysosomal Therapeutics in Cambridge, Massachusetts, are developing drugs for Parkinson’s that target GBA. Researchers in Canada recently evaluated the ability of the chaperone ambroxol to stimulate GBA activity in a small Phase 2 Parkinson’s study, and a similar trial of ambroxol is starting up in London. Amicus Therapeutics, Cranbury, New Jersey, is developing the small-molecule GBA chaperone AT3375 for both Gaucher’s and Parkinson’s diseases.—Madolyn Bowman Rogers





Comments
No Available Comments
Make a Comment
To make a comment you must login or register.