At the 10th Brain Research Conference: RNA Metabolism in Neurological Disease, a satellite of the Society for Neuroscience annual meeting, attendees learned about a parade of mice modeling different aspects of ALS and FTD that result from expansions in the C9ORF72 gene. Knockouts lose control of their immune systems, while transgenics overexpressing the repeat-heavy human gene ranged from normal to having movement and cognitive abnormalities. The results suggest the repeats cause toxicity, but scientists have plenty more to do to understand what the mice are telling them.
C9ORF72 Mice Point to Gain of Toxic Function in ALS, FTD
Over the past several years, much research has pointed to cellular mishandling of RNA as a key factor in neurodegeneration. Numerous RNA-binding proteins—from the well-known TDP-43 and fragile X mental retardation proteins to relative newcomers such as hnRNPA3 and RBM45—mutate or aggregate in diseases such as amyotrophic lateral sclerosis, frontotemporal dementia, and myotonic dystrophy. As a result, scientists believe RNAs may be misspliced, sequestered from the ribosome, or otherwise fouled up. This convergence of mechanisms made the RNA Metabolism in Neurological Disease conference, held October 15-16 in Chicago before the Society for Neuroscience annual meeting, the place to be for scientists interested in ALS, FTD, and similar disorders. Attendees were astounded by the latest news about RNA-binding proteins and C9ORF72, a gene with ALS- and FTD-linked repeat expansions that encode anomalous repetitive RNAs and peptides. Co-organized by Paul Taylor of St. Jude Children’s Research Hospital in Memphis, Tennessee, and Fen-Biao Gao of the University of Massachusetts Medical School in Worcester, the meeting featured new mouse models of C9ORF72 disease. Scientists who eliminated the gene from their animals noticed powerful inflammation, while those who expressed a mutant transgene with repeats detected cognitive and motor defects.
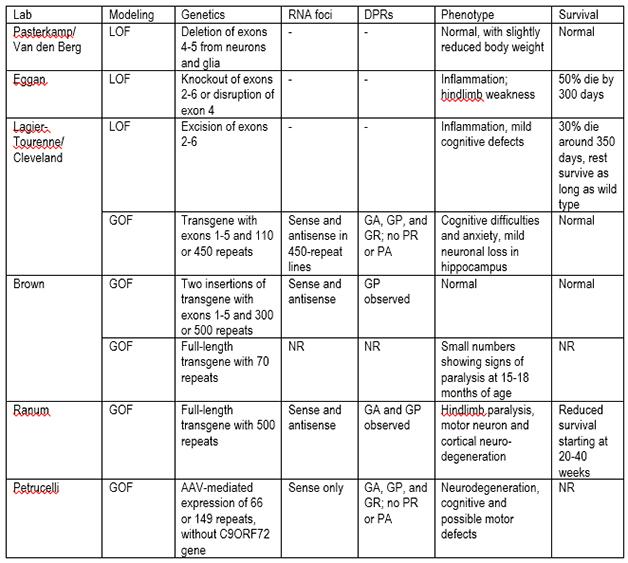
C9ORF72 Mouse Models
C9ORF72 model mice. LOF=loss of function; GOF=gain of function; DPRs=dipeptide repeats; NR=not reported; GA=glycine-alanine; GP=glycine, proline; GR=glycine-arginine; PR=proline-arginine; PA=proline-alanine.
C9ORF72 encodes a protein of uncertain function. Hexanucleotide repeats in the gene’s first intron—numbering in the hundreds or thousands—have been linked to ALS and FTD (see Sep 2011 news). Cells transcribe these repeats into messenger RNAs in both the sense and antisense directions, and these RNAs aggregate into nuclear foci (see Jan 2013 conference news; Nov 2013 news). Despite being in introns, the repeats somehow avoid getting spliced out of mRNAs—scientists are not yet sure how—and ribosomes then translate them through a process known as repeat-associated non-ATG translation. Five different repeat dipeptides result from the six possible reading frames in the sense and antisense directions (two reading frames encode the same dipeptides). These peptides can gather into toxic aggregates (see Feb 2013 news).
Researchers are trying to work out how the repeats instigate disease by differentiating among three hypotheses. For one, the repeats interfere with normal C9ORF72 production, leading to a loss of function when people only make half the normal amount of the protein. Two, the RNA foci might cause a gain of function that damages neurons. Three, the translated dipeptides might be toxic. Several research groups attempting to model these scenarios in mice presented their preliminary results in Chicago (see table above). The upshot: No model quite mimics the human condition, said Robert Brown of the University of Massachusetts Medical School in Worcester, who has engineered two transgenic lines. With multiple mouse lines still under study, it is too soon to draw firm conclusions about ALS and FTD from the animals, said Clotilde Lagier-Tourenne of Massachusetts General Hospital in Charlestown, who is analyzing both loss-of-function and gain-of-function models. Nonetheless, researchers at the meeting seemed confident that these mice will offer clues about pathology. They will also serve as testing grounds for therapeutics, such as antisense oligonucleotide therapy to eliminate repeat RNAs and their protein products. “We are making lots of progress building models for C9ORF72,” said Lagier-Tourenne. “It is very exciting.”
Knocking Out C9ORF72 Kicks Up Cytokine Storm
Two groups addressed the loss-of-function idea by generating C9ORF72 knockout mice. Kevin Eggan and colleagues at Harvard University, and a collaboration between Lagier-Tourenne’s group and Don Cleveland’s lab at the University of California, San Diego, generated mice from the same embryonic stem cell line that lacks exons 2–6 of the 12 found in the endogenous C9ORF72 gene. Mice heterozygous for the truncated gene made 50 percent of the normal amount of C9ORF72 RNA and protein. Homozygous mice completely lacked both transcript and protein.
The homozygote knockouts exhibited problems reminiscent of ALS and FTD. Eggan said their animals developed weakness in their hind legs and struggled to breathe as they aged. Lagier-Tourenne said their mice had no obvious motor defects but developed mild behavioral abnormalities at six months. They erred navigating mazes and the males were highly anxious, said Jie Jiang of UC San Diego, who presented the data on a poster.
Eggan’s group used electromyography to detect denervation at the neuromuscular junction in their animals, but came up with baffling results. They found denervation and reduced motor neuron counts in the first group of animals they assessed, but not in the next, supposedly identical cohort. He has not yet figured out why. The knockouts died early. Jiang reported that about 30 percent of his mice died after around a year, while less than half of Eggan’s mice made it to their first birthday.
While the Cleveland/Lagier-Tourenne heterozygotes survived normally, the Eggan heterozygotes exhibited a diluted version of the homozygous phenotype, and some of them died within a year as well. The differences between the populations might be simply due to the animals’ environments, Jiang said, noting that mice are quite sensitive and even something like construction noise outside could impact their phenotype.
Eggan noted that the mice died from a variety of causes. Some suffered respiratory failure. Others exhibited such severe loss of motor control or skin problems that veterinarians recommended they be sacrificed. Some were found, on necropsy, to have bled internally. The most striking change, Eggan said, was grossly enlarged spleens in the homozygous C9ORF72 knockouts (see image below). Many mice also had swollen lymph nodes in their necks. Lagier-Tourenne and colleagues noticed enlarged spleens and lymph nodes in their animals, as well.
Immune indicator
C9ORF72 knockout mice possessed much larger spleens (right) than normal mice (left), suggesting inflammation. [Image courtesy of Aaron Burberrry, Harvard University.]
The enlarged organs indicated defects in the body’s blood-cell supply, which Eggan’s group pursued further. The animals produced slightly lower red blood cell counts and fewer platelets than normal, but an excess of white blood cells. These findings made the researchers suspect an imbalance in cytokines, immune signaling molecules. Indeed, numerous cytokines, including interferon-γ and IL-5 were upregulated, mimicking a hyper-immune reaction sometimes termed a “cytokine storm,” which can be fatal in humans. The blood and cerebrospinal fluid of people with ALS also contains upregulated cytokines—IL-17 and IL-23—Eggan noted, though he said the cause may be different (Rentzos et al., 2010).
To determine if the mouse defects resulted from C9ORF72 loss in the hematopoietic system, Eggan’s group transplanted bone marrow from C9ORF72 knockouts into wild-type mice and vice versa. One of the first things they noticed was that wild-type animals, upon receiving knockout bone marrow, developed bulky chests due to swollen lymph nodes beneath their skin. Their spleens grew large, though not as large as those from full C9ORF72 knockouts. Their lifespan was shortened, though not as much as complete knockout mice. Transplanting wild-type bone marrow into knockout mice partially rescued the enlarged spleen phenotype, and enhanced survival.
Eggan’s group used CRISPR gene editing to generate a separate line of mice (see Sep 2014 series) with a premature stop codon in exon 4. The researchers have not yet fully evaluated the RNA and protein levels in these mice, but already know that they, too, have anemia, cytokine storm, and enlarged spleens. As with the other knockout lines, many died before a year of age.
The signs are not reminiscent of neurodegeneration but of blood cancer, commented Jeffrey Rothstein of Johns Hopkins University in Baltimore. Lagier-Tourenne said pathologists told her the mice appeared to have lymphoma. However, the cancer experts Eggan consulted told him he was seeing something else. “It’s more of a massive inflammatory condition than cancer,” he said. He speculated that inflammation might be behind some motor deficits of the knockouts, such as falling off a rotating rod. He quipped that he, too, might drop off a rotating rod if he were weathering a cytokine storm.
Dieter Edbauer of Ludwig-Maximilians University of Munich agreed that these mice indicate that C9ORF72 performs some function in the immune system. However, he, Jiang, and Eggan pointed out that knockout mice with no C9ORF72 are not directly comparable to people with the C9ORF72 repeat expansion because the people make some C9ORF72 protein from their one good gene. At least, Jiang said, the mice allow him to conclude that loss of C9ORF72 function by itself does not precipitate neurodegeneration. In this he agrees with the authors of a recent paper who observed no signs of neurodegeneration or change to mortality in mice missing C9ORF72 only in their nervous system. Those mice had normal levels of the gene in the rest of the body, which presumably precluded the inflammatory blood phenotype (see Jun 2015 news).
Wrangling Repeats into Mice Yields Conflicting Results
What, then, happens to transgenic mice modeling gain of function? The first published gain-of-function model, discussed by Leonard Petrucelli of the Mayo Clinic in Jacksonville, Florida, relies on AAV-mediated expression of only the hexanucleotide repeat in the brain and spinal cord. These mice, expressing 66 repeat copies, mimic several characteristics of human disease: RNA foci and dipeptide inclusions as well as TDP-43 aggregates in the brain, plus motor and cognitive symptoms (see May 2015 news). Petrucelli’s lab is now working on a new version with 149 repeats, with which they have already detected poly-glycine-proline dipeptides, he said.
Other scientists created traditional transgenic mice, starting with human C9ORF72 genes copied into bacterial artificial chromosomes (BACs), then dropped into the mouse genome. Both the Brown lab and the Cleveland/Lagier-Tourenne collaboration started with the same partial C9ORF72 construct, comprising the gene’s first five exons and including a 450 hexanucleotide expansion. However, because the guanine- and cytosine-rich repeats are unstable, the expansion can grow or shrink in bacterial hosts. The upshot was that the mice had different repeat numbers, though once in a mouse the given sequence length stays stable, Jiang noted.
He and Lagier-Tourenne said they managed to create several mouse lines, selecting four for detailed analysis. One line carried a transgene with 110 repeats, and three contained a gene with 450 repeats though expressed at different levels. This panel, including both homozygous and heterozygous BACs, allowed the researchers to measure pathology at varying doses of the transgene. The line they have best characterized has 450 repeats and the transgene is expressed at about 3.5 times endogenous C9ORF72 levels, Jiang told Alzforum. Meanwhile, Brown’s lab obtained one stable mouse line with two transgene insertions in the genome, one with 300 repeats and the other with 500, yielding four total transgenes in the homozygotes. The researchers analyzed animals with transgene expression similar to that of the endogenous gene, Brown said.
Brown and colleagues observed a slight decrease in motor neuron numbers compared to control mice, but it was not statistically significant, he said. The scientists saw no evidence of denervation at the neuromuscular junction. Lagier-Tourenne reported that the lines with 450 repeats, at 12 months, exhibited mild loss of hippocampal neurons.
As suspected, the C9ORF72 pathology was dose-dependent, in terms of both repeat number and expression level. RNA foci, both sense and antisense, appeared in mice with 300 to 500 repeats, but not mice with only 110 repeats. Animals with greater transgene expression exhibited more foci as well. Cleveland and Lagier-Tourenne also detected the three dipeptide repeats translated from the sense C9ORF72 RNA, poly-glycine-alanine, poly-glycine-proline, and poly-glycine-arginine. Using an immunoassay specific for glycine-proline, both groups detected soluble dipeptide repeats in the brains of young mice. The soluble peptide transitioned to immunoreactive aggregates in older animals, Lagier-Tourenne said.
Though the mice exhibited these molecular signs of C9ORF72 disease, there was little wrong with them physically. The Lagier-Tourenne/Cleveland mice moved normally, though by one year of age they struggled to navigate mazes, and exhibited mild neurodegeneration in the hippocampus. They lived normal lifespans. Brown’s animals, now two years old, are of normal weight and without motor defects. They were also cognitively normal in behavioral tests including marble burying—anxious mice entomb the unfamiliar objects—and dominance interactions between old and young mice, Brown reported. Those animals carried just the first five exons of the C9ORF72 gene.
In addition, Brown’s group generated a mouse line that contains the full-length C9ORF72 transgene, albeit with only 70 repeats. The researchers have not analyzed the animals thoroughly yet, but for now Brown reported that some of them are starting to exhibit paralysis at 15 to18 months of age.
Laura Ranum of the University of Florida in Gainesville, also generated mice that contain full-length C9ORF72 transgene, with 500 repeats. Some of the mice died between 20 and 40 weeks of age, their demise preceded by weight loss, failing grip strength, and inability to move their hind legs. Others survived longer, but by one year, 80 percent had died or developed phenotypes such as anxiety and a hunched back typical of some other ALS models. The scientists observed neuron loss consistent with both ALS, with fewer motor neurons in the spinal cord and denervation at neuromuscular junctions, and FTD, with neurodegeneration in the cortex. The animals also accumulated sense and antisense RNA foci in their brains and spinal cords, as well as aggregated dipeptide repeats.
“All these mice suggest that the repeats have a gain–of-function toxicity,” Jiang concluded. However, researchers were left scratching their heads about the variation between the lines. The data so far seem to indicate that the full-length C9ORF72 transgene might somehow be more toxic than the partial construct, but researchers need to compare notes, Brown said. Lagier-Tourenne cautioned that scientists check the sequences of their constructs carefully. She speculated that extra genes contained in the short or long BAC clones, including possibly unannotated ones, might be responsible for some phenotypes. Taylor also theorized that something in the larger transgene might influence transcription levels for the antisense RNAs. “We are not at the end of the road yet,” he said.
Indeed, Lagier-Tourenne and Cleveland are already plotting their next mouse line, a cross between their heterozygous C9ORF72 knockouts and the transgenics with the repeats. This, they hope, will yield an animal more representative of human C9ORF72 expansion carriers, who have half the normal complement of the normal gene along with the repeat-expanded version.—Amber Dance
Does C9ORF72 Repeat RNA Promote Protein Phase Transitions?
Over the past couple of months, scientists have learned that RNA-binding proteins such as FUS and hnRNPA1 undergo phase transitions, separating themselves and their associated nucleic acids and protein partners into liquid-phase-like organelles that are distinct from the surrounding cytoplasm or nucleoplasm (see today’s Alzforum Webinar). At the third biennial RNA Metabolism in Neurological Disease meeting, held in Chicago October 15-16, researchers from Brigham and Women’s Hospital in Boston offered preliminary data indicating that RNAs themselves might seed formation of these liquid organelles, too. It looks as if RNAs might manage the same trick that previously only proteins were thought to perform, said Marta Fay, who presented the work in a poster. The meeting was a satellite to the Society for Neuroscience meeting.
Pernicious seed. C9ORF72 RNAs form G-quadruplexes that attract RNA-binding proteins, leading to large accumulations that might take the form of aggregates or hydrogels. [Courtesy of Marta Fay, Brigham and Women’s Hospital.]
Fay, who works in the laboratory of Paul Anderson, and Brigham collaborator Pavel Ivanov studied RNA from C9ORF72, a gene implicated in amyotrophic lateral sclerosis and frontotemporal dementia. Carriers of the expanded gene often express hundreds or thousands of GGGGCC repeats in their RNA, as well as CCCCGG repeats made from transcription in the antisense direction (see Sep 2011 news; Jan 2013 conference news; Nov 2013 news). Scientists suspect the repetitive RNAs, which assemble into foci, could be toxic. Might they cause problems by inducing liquid- or solid-phase transitions in the cell?
If so, Fay would expect to see the C9ORF72 RNAs associating with components of liquid organelles such as stress granules. She incubated the repeat RNAs with lysates from mouse brain or cultured U2OS cells, a commonly used human osteosarcoma line in which stress granules have been studied in the past. Once Fay added the C9ORF RNAs, she observed the formation of precipitates. Mass spectrometry identified 187 molecules in them, including known stress granule constituents such as G3BP1, eIF3b, mRNAs, and ribosome subunits. Other precipitated proteins are not typically found in stress granules, but some normally make up the related P bodies, suggesting the C9ORF72 RNAs associated with the members of multiple types of RNA granules.
As a control, Fay compared her precipitation process to that induced by biotinylated isoxazole (b-isox), a chemical known to shepherd RNA granule components into a hydrogel (Kato et al., 2012). C9ORF72 RNA acted like b-isox, precipitating the RNA-binding proteins in a temperature- and concentration-dependent manner.
These precipitates only formed when Fay added GGGGCC. The antisense CCCCGG RNA yielded no aggregation. The sense repeats are known to assemble into structures called G-quadruplexes, where G-rich sequences within a single RNA strand line up like the four sides of a building (see image above). Fay thought this might explain why only the sense sequence precipitates RNA granule components. It did. When Fay added drugs that promoted G-quadruplex formation, she saw more precipitates. When she applied treatments that dissolved the quadruplexes, she obtained less.
Finally, Fay tested her ideas in cells, again from the U2OS line. Expressing the repeat RNA in those cells induced formation of stress granules, and the longer the repeats, the more granules appeared. She proposed that the extensive C9ORF72 repeats might over-stabilize granules, pushing the proteins therein toward pathological fibrilization (see image above).
Researchers are not sure yet precisely what these precipitates are, and if they are liquid, gel-like, or solid in nature. Paul Taylor of St. Jude Children’s Research Hospital in Memphis, Tennessee, a meeting co-organizer and panelist at the Alzforum Webinar, thought Fay’s results were not necessarily indicative of phase separation. Ivanov said he and his colleagues would collaborate with biophysicists to solve the question.
Supporting Fay’s findings, other scientists have discovered that RNAs are not merely caught up in granule organelles, but actively participate in the phase transition. “RNA can impact both the assembly of droplets and their properties,” commented Cliff Brangwynne of Princeton University, a pioneer of the field of protein phase transition. Brangwynne did not attend the Chicago meeting but will lead off today’s Webinar. “RNA can also influence the viscoelastic properties of the droplets,” he told Alzforum (Berry et al., 2015; Elbaum-Garfinkle et al., 2015; Zhang et al., 2015).—Amber Dance
RNA Regulator Locked Out of Nucleus by C9ORF72 Repeats
Cells possess a protein that can alleviate C9ORF72 repeat toxicity—ironically, it may not be able to reach the nuclear RNA repeats to silence them. Christian Haass of the German Center for Neurodegenerative Diseases (DZNE) in Munich has proposed that the RNA-binding protein hnRNP A3 dismantles the repetitive RNAs associated with some cases of amyotrophic lateral sclerosis and frontotemporal dementia. Heterogeneous nuclear ribonucleoprotein A3 must enter the nucleus to clear the repeat RNAs, but they, in turn, interfere with normal nucleocytoplasmic transport. HnRNP A3 represents the first reported victim of this defective nuclear entry, Haass told Alzforum. He presented his data at the RNA Metabolism in Neurological Disease meeting held October 15-16, a satellite of the Society for Neuroscience annual conference in Chicago.
Double trouble. Poly-dipeptides (red) encoded by the C9ORF72 repeat, and aggregates of p62 (green), a common marker of ALS inclusions, co-localize (right) in HeLa cells. [Courtesy of Christian Haass, DZNE.]
The C9ORF72 gene, which encodes a protein of as-yet-undetermined function, normally contains up to 23 hexanucleotide repeats. That number can grow into the hundreds or thousands, and when it does, it amplifies risk for ALS and FTD. Scientists are not yet sure how the repeats cause disease, but the sequences are abnormally transcribed into RNAs that are translated into poly-dipeptide repeats (DPRs). These RNAs and DPRs aggregate into foci and inclusions, respectively. Scientists recently reported that either the RNAs or DPRs, or both, interfere with protein trafficking across the nuclear pore (see Aug 2015 news).
After C9ORF72 repeats were linked to neurodegeneration (see Sep 2011 news), Kohji Mori in Haass’ lab went hunting for RNA-binding proteins that might interact with the repeat RNA. He identified heterogeneous nuclear ribonucleoprotein (hnRNP) A3, an RNA transporter (see Feb 2013 news). This hnRNP possesses sequences similar to hnRNP A1 and hnRNP A2/B1, which are mutated in rare ALS cases and also bind C9ORF72 repeat RNAs (see Mar 2013 news). Scientists have not yet discovered hnRNP A3 mutations in ALS, but Haass predicts they will (Calini et al., 2013).
As Mori and Haass examined autopsy tissues from C9ORF72 repeat carriers, they noticed that hnRNP A3 was often absent from the nucleus, where it normally resides. It also turned up in aggregates in both the nucleus and cytoplasm, and occasionally co-localized with p62, a common component of pathological inclusions in ALS. The researchers suspected that since it was mislocalized, hnRNP A3 failed to perform its normal function in the C9ORF72 patients.
To test the effects of hnRNP A3 loss of function in the lab, Mori used small interfering RNAs to knock down hnRNP A3 in HeLa cells expressing 80 copies of the C9ORF72 hexanucleotide repeat. These cells generate repeat RNA foci and produce repeat poly-dipeptides, which sometimes aggregate with p62 (see image above). In the hnRNP knockdowns, the results were striking. The repeat RNA shot up to much higher levels. “It was a day-and-night difference,” Haass told Alzforum. Without hnRNP A3, the cells overproduced the repeat RNA by six- or sevenfold, he said, and accumulated many more RNA foci. The cells also made much more of the repeat poly-dipeptides than control cells did. Mori and collaborator Dieter Edbauer of DZNE obtained similar results when they knocked down hnRNP A3 in primary rat neurons.
If loss of hnRNP A3 allowed C9ORF72 repeats to run amok, then augmenting expression of the RNA-binding protein would tone that down, the authors predicted. Sure enough, overexpressing hnRNP A3 in HeLa cells diminished both repeat RNA and poly-dipeptide proteins.
How does hnRNP A3 work? Because C9ORF72 repeat RNA folds back on itself in tight hairpins, the researchers suspected hnRNP A3 altered this conformation. Haass that suggested hnRNP A3 binds to the repeats, unwinding their secondary structure and exposing them to degradation by RNases. Consistent with this, hnRNP A3 sans RNA-binding domain failed to attenuate C9ORF72 pathology in HeLa cells lacking the normal hnRNP A3 gene.
Back when they looked at human tissues, Mori and Haass had noticed that hnRNP A3 was missing from the nucleus, so they predicted it might be unable to protect cells from repetitive RNAs if it was mislocalized. To find out, Mori removed the nuclear localization signal from the hnRNP A3 gene and expressed it in their modified HeLa cells. Compared with the wild-type gene this construct weakly rescued cells from C9ORF72 pathology, reducing repeat RNAs and poly-dipeptides by about half. Haass attributed the partial rescue to some hnRNP A3 that made it into the nucleus by chance, which he said often occurs with nuclear proteins lacking their nuclear localization sequence.
Together, these findings led Mori and Haass to propose that hnRNP A3 represses expression of C9ORF72 repeats, but only if it can get into the nucleus. They predicted that less hnRNP A3 in the nucleus would spell more C9ORF72 repeat pathology. The scientists tested this in human autopsy tissues. So far, they have examined the brains of 18 people who died of C9ORF72-based ALS-FTD. Mori calculated the ratio of hnRNP A3 to poly-glycine-alanine inclusions in individual neurons. Indeed, the less hnRNP A3 he observed in the nucleus, the more polyGA pathology in the cell.
“This is interesting because they identified an RNA-binding protein that suppresses DPR protein production,” commented Fen-Biao Gao of the University of Massachusetts Medical School in Worcester, who co-organized the conference. Gao told Alzforum he found the results from human tissues to be particularly striking.
C9ORF72 repeats could instigate a vicious cycle, Haass theorized. Once a few escape the nucleus and interfere with nucleocytoplasmic transport, hnRNP A3 could get stuck out in the cytoplasm. It would be unable to unwind newly transcribed C9ORF72 RNA, allowing the concentration of repeat RNAs and dipeptides to skyrocket and further blocking entry of hnRNP A3, and other proteins, to the nucleus.—Amber Dance
Listen Up, Gene Silencing Strikes a Chord at RNA Meeting
Scientists are taking the offensive against neurodegeneration, with tiny nucleic acids as one of their favored weapons. RNA Metabolism in Neurodegenerative Disease, an SfN satellite conference held October 15-16 in Chicago, hummed with excitement about progress toward antisense oligonucleotide (ASO) therapies that would shut down translation of genes involved in frontotemporal dementia, amyotrophic lateral sclerosis, and Huntington’s disease. “So far all signs are quite positive for ASOs for C9ORF72, SOD1, and huntingtin,” commented Paul Taylor of St. Jude Children’s Research Hospital in Memphis, Tennessee, who co-organized the symposium. Trials are already ongoing or planned for Huntington’s ALS.
ASOs are just one way to get rid of problem RNAs and proteins. Meeting attendees were also intrigued by novel methods to prevent transcription or refold toxic proteins. Aaron Gitler of Stanford University in Palo Alto, California, claimed that a protein required to transcribe trinucleotide and hexanucleotide repeats might make a good drug target. If scientists could suppress it, they might mitigate the production of toxic repetitive RNAs and any downstream protein products, he said. In addition, James Shorter of the University of Pennsylvania Perelman School of Medicine in Philadelphia shared his latest data on augmenting a chaperone that reshapes a variety of malformed proteins involved in neurodegeneration.
Little RNA, big effects.
Robert Brown hopes to use microRNAs to attenuate expression of toxic SOD1 and C9ORF72. [Courtesy of Paul Gardner, University of Canterbury, Christchurch, New Zealand.]
Sensible Strategy
The concept of using ASOs to shut down unwanted protein translation has been around for decades, but finally saw some success in recent years with the 2013 approval of a whole-body ASO treatment, Kynamro®, to treat a genetic cholesterol disorder. Kynamro’s maker, Isis Pharmaceuticals of Carlsbad, California, collaborates with several scientists who hope ASOs will vanquish neurodegenerative disease. Isis’ expertise in designing and manufacturing large quantities of ASOs allows researchers to test hundreds of versions before they pick the best choice for a trial, noted Don Cleveland of the University of California in San Diego. Isis also collaborates with many scientists on preclinical and clinical studies. At the meeting, Cleveland reported on the latest progress for ASOs in animal models as well as human trials.
Cleveland has been working with Isis for 10 years with the goal of ASO treatments for neurodegenerative disease. Some scientists were skeptical about the idea, he told Alzforum, but the researchers determined it was a feasible strategy with a joint UCSD-Isis study of ASOs for the ALS gene SOD1. It slowed down the disease in transgenic mice (see Jul 2006 news). Further, anti-SOD1 ASOs sailed through their first human safety trial with no signs of ill effects (see Jan 2013 conference news; May 2013 news). Now, the company has a second-generation, more potent SOD antisense oligo to try. It has partnered with Biogen Idec of Cambridge, Massachusetts, to run a Phase 1 study. Biogen is still finalizing the details, a representative told Alzforum, and expects the study to commence in early 2016.
Isis is already in the clinic with a Phase 1/2 trial, started in July, of antisense therapy against huntingtin, the gene mutated in Huntington’s disease. It too delivered promising preclinical results, delaying progression and reversing symptoms in mice (see Jun 2012 news). In the human trial, 36 participants will receive either the ASO or a placebo in a double-blinded fashion, in four infusions over about 13 weeks. Those in the active treatment arm will receive escalating doses. Though safety is the primary outcome measure, the researchers will also assess cognitive performance. Researchers expect the study to wrap up in the fall of 2017.
Cleveland is now targeting C9ORF72, a gene that can contain a large repeat expansion that raises risk for ALS and FTD. The scientists designed an ASO that targets only the repeat-containing RNA, and confirmed that it left the normal gene unperturbed. “This is almost allele-selective silencing, the sort of Holy Grail in these kinds of gene-silencing approaches,” Cleveland said. Cleveland, along with Clotilde Lagier-Tourenne of Massachusetts General Hospital in Charlestown, tested the antisense therapy in new mouse models of C9ORF72 toxicity they developed (see Part 1 of this series). They treated four to seven mice with a single ASO injection into the brain’s ventricles. Two and four weeks later, when the researchers sacrificed the mice, the animals had fewer repeat-containing mRNAs in the brain and spinal cord than mice treated with control, ineffective ASOs. The treatment also reduced levels of at least two of the poly-dipeptides encoded by those repeats, poly-glycine-proline and poly-glycine-alanine. The researchers have not yet measured the other peptides. They plan to check if the treatment alleviates learning defects in these mice, as well, said Jie Jiang of Cleveland’s lab, who presented the project in a poster. Cleveland predicted C9ORF72 ASOs would be ready for a human trial in early 2017.
Micro-Managing Robert Brown of the University of Massachusetts Medical School in Worcester prefers a different approach to rid neurons of unwanted RNAs. MicroRNAs are natural, noncoding sequences that bind to target mRNAs to fine-tune gene expression, by either preventing translation or promoting mRNA degradation. Brown and colleagues have already engineered an artificial miRNA against SOD1, which they packaged into an adenoassociated virus vector. They injected the vector into the spinal cord of two-month-old transgenic mice expressing a human SOD1 with the glycine-93-alanine mutation that causes ALS. The virus delivered that miRNA gene to the nucleus, so the neurons generated the miRNA themselves after the one-time treatment. It knocked down the mutated gene and slowed the disease (Wang et al., 2014). Now, the lab is using the same strategy to suppress C9ORF72.
Brown’s group has tried the miRNAs on cultured cells, and also provided them via virus to a mouse model they engineered to overexpress the repeat-laden human C9ORF72 gene. The researchers are experimenting with intrathecal, intraventricular, and even intravenous delivery, he said, since some viruses can cross the blood-brain barrier. They have treated both newborns and adults. “We see promising results,” Brown told Alzforum. The repeat RNAs formed fewer potentially toxic foci, and expression of poly-dipeptides fell.
How do Brown’s microRNAs compare to Cleveland’s ASOs? Each has its pluses and minuses, Brown said. Unlike viruses, most ASOs cannot cross the blood-brain barrier. AAV delivers a permanent transgene, so it would require only one treatment as opposed to regular infusions of ASOs. However, that means there would be no way to take back a viral microRNA treatment gone awry. Brown also noted that microRNAs mature in the nucleus and then find their target mRNAs in the cytoplasm. Scientists recently determined that C9ORF72 repeats inhibit nucleocytoplasmic transport, which Brown speculated might interfere with the transport of miRNAs to the cytoplasm (see Aug 2015 news). Brown’s group is exploring whether traffic jams at the nuclear membrane will interfere with their microRNAs, he told Alzforum.
Transcription Factor
In the case of C9ORF72, treatment with ASOs and miRNAs faces another complication. Because the cell transcribes the repeats in both the sense and antisense directions, oligonucleotides specific for the forward and backward directions may be needed to treat the disease (see Nov 2013 news). Gitler proposed a potential solution that would eliminate both antisense and sense transcripts at once.
A few years ago, scientists discovered that the yeast transcription elongation factor SPT4 assisted RNA polymerase in copying lengthy trinucleotide repeats such as those found in mutant huntingtin (see Feb 2012 news). SPT4—and its mammal homolog Supt4h—help the polymerase stay attached to the repetitive DNA, but are not essential for most transcripts or cell viability (Malone et al., 1993). Since then, the same group has knocked down Supt4h in HD model mice, and found it diminished expression of huntingtin RNA and protein (Cheng et al., 2015). Might SPT4 also assist RNA polymerase transcribing C9ORF72 hexanucleotide repeats? Gitler’s group found that if they eliminated SPT4 from yeast, C9ORF72 repeats were no longer transcribed or translated. He plans to test if downregulating Supt4h protects cell lines derived from people with C9ORF72 expansions, or mice that model C9ORF72 toxicity.
Optimized Chaperone
While Gitler, Brown, and Cleveland have prevention of translation in mind, Shorter has devised a solution to problem proteins once they are out and about, and misfolded as toxic neurodegeneration proteins often are. The researchers started with Hsp104, a chaperone known to disassemble oligomers. Using error-prone PCR, they generated several Hsp104 variants that protect yeast from toxic proteins. The modified proteins untangled and refolded mutant forms of α-synuclein, TDP-43, FUS, and TAF15 into their proper conformations (see Aug 2014 news). At the meeting, Shorter added to that list the ribonucleoprotein coded by the ALS gene hnRNPA2, and a poly-dipeptide (poly-proline-arginine) encoded by C9ORF72 repeats. “These [chaperones] are the only things we know about that are able to separate aggregated proteins and recover native forms,” Shorter said, adding he would love to do the same in people with ALS.
Shorter shared some preliminary data to suggest that chaperones can act as disaggregases in animals. With his modified Hsp104, he was able to dissolve aggregated SOD1 in nematodes and globs of FUS in cultured human fibroblasts, he said. He has yet to test the modified chaperone in mice. While people have no Hsp104 homolog, Shorter speculated that he might be able to modify a different human chaperone to make it disaggregate toxic proteins. He envisions either providing the protein as a therapeutic, or the gene via gene therapy. The idea of disassembling aggregates has appeal, commented Robert Bowser of the Barrow Neurological Institute in Phoenix. However, he added, any such therapeutic would likely require tight control so it would not unwind essential proteins.
Cleveland hinted he is already looking ahead to the next big thing, which he predicted would be CRISPR-based gene editing (see Sep 2014 series). “You could even imagine getting gene correction,” he said. Brown told Alzforum exciting times are ahead. “Gene silencing and gene replacement are finally coming of age.”—Amber Dance
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu XD, Bennett CF, Cleveland DW, Ravits J.
Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration.
Proc Natl Acad Sci U S A. 2013 Nov 19;110(47):E4530-9. Epub 2013 Oct 29
PubMed.
Stressed Out: RNA-binding Protein Inhabits Granules in ALS, FTD
Panning the cerebrospinal fluid of people with amyotrophic lateral sclerosis, researchers discovered a new pathological marker in RNA-binding motif 45 (RBM45). In diseased neurons, this protein hangs out in cytoplasmic stress granules, a familiar compartment for scientists interested in ALS. It also occupies nuclear stress bodies, a new structure for the field. “How the nucleus responds to stress has been unexplored territory for ALS and other neurodegenerative disease,” said Robert Bowser of the Barrow Neurological Institute in Phoenix, who presented the work at RNA Metabolism in Neurological Disease, a Society for Neuroscience satellite symposium held October 15-16 in Chicago.
RBM45 Domains.
[Courtesy of Li et al., Scientific Reports.]
Like the well-known ALS proteins TDP-43 and FUS, RBM45 contains RNA-binding domains. While it lacks the prion-like low-complexity sequences of the other two proteins, it sports a nuclear localization sequence, as FUS does (see image above). Though no current evidence indicates genetic variants near the RBM45 gene associate with ALS or any other neurodegenerative disease, Bowser and collaborators are checking sequences of people with familial ALS.
Scientists know little about RBM45. It is expressed highly in the brain and upregulated following spinal-cord injury (Mladinic et al., 2010). Bowser and colleagues observed that while the protein appears to be predominantly nuclear, some collects in the cytoplasm. In both cellular locations, it makes a speckled pattern consistent with the large protein/RNA complexes formed by RNA-binding proteins. Bowser suspects it may shuttle between the two compartments. RBM45 also condenses into cytoplasmic inclusions in the motor neurons of ALS patients. There, it co-localizes with TDP-43 and ubiquitin, common pathological markers of the disease. It also forms cytoplasmic inclusions in people who died of frontotemporal lobar degeneration or Alzheimer’s disease (Collins et al., 2012).
A New, Nuclear Stage for Pathology
Bowser reported in Chicago that the RBM45 also formed nuclear inclusions. These were much smaller than typical cytoplasmic stress granules, Bowser said, and did not co-localize with TDP-43 or ubiquitin, common markers for pathological aggregates in the cytoplasm. They formed in both neurons and glia. In autopsy samples of hippocampi and spinal cords, the average nucleus contained about three of these puncta. In ALS, FTD, and AD cases, there were about 10.
What were these inclusions? Testing both neural and non-neuronal cell lines with antibodies to various nuclear substructures, Bowser discovered the puncta contained the heat shock transcription factor 1 (HSF1). In addition to activating genes in response to high temperatures, HSF1 helps generate nuclear stress bodies. Very little is known about these structures, but they are believed to protect against stress by recruiting transcription and splicing factors to DNA to modulate gene expression, said Bowser. Notably, nuclear stress bodies only occur in primate cells, he said.
To see if nuclear stress bodies are important in disease states, the researchers re-examined the autopsy tissue. They found more puncta containing scaffold attachment factor B (SAFB), another stress body marker, in the ALS and FTD cases than in the controls. Now, the scientists are working to understand the function of nuclear stress bodies and how they act on gene expression in chronically stressed neurons during neurodegeneration, Bowser told Alzforum.
Plenty of Action in the Cytoplasm
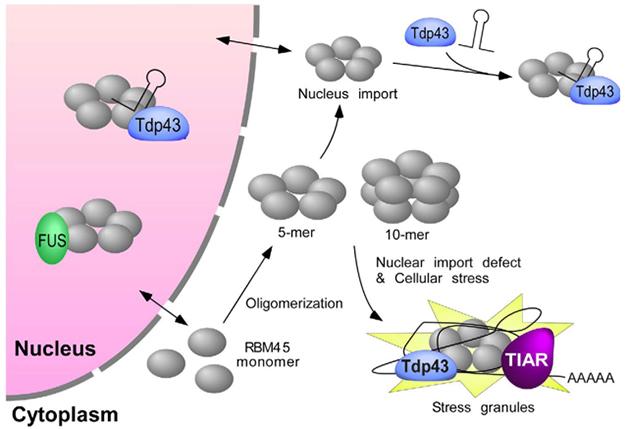
Even as they analyze the RBM45-containing stress bodies, Bowser’s group is working to understand the protein and its interactions at a molecular level. In a poster, Yang Li reported that individual RBM45 proteins associate in multimers. Li got hints of this first from co-immunoprecipitation experiments and then by using a chemical cross-linker to stabilize multimers. In the presence of the cross-linker, RBM45 migration in polyacrylamide gels corresponded to that of a pentamer. RBM45 might form a ring, Li speculated, and two rings might stack to form a decamer. Using a series of deletion mutants, she determined that the mid-section of the protein, which she named the homo-oligomer assembly (HOA) domain, was necessary for this oligomerization (see image above).
RBM45 interactors. RBM45 monomers assemble into multimers that associate with TDP-43 via a bridging RNA. Multimers may end up in stress granules in the cytoplasm, or stress bodies in the nucleus. [Courtesy of Li et al., Scientific Reports.]
What about interactions with other proteins? Li has not yet examined nuclear stress bodies but did study how RBM45 interacts with TDP-43 in the cytoplasm. The two proteins co-immunoprecipitated—but not, however, if Li treated the cells with RNase. She thinks they may bind and co-regulate the same transcripts. Because RBM45 also needed the HOA to associate with TDP-43, Li thinks RBM45 must bind RNAs as an oligomer. The researchers are now trying to identify the RNAs that bind both RBM43 and TDP-43 (Li et al., 2015).
In another recent paper, Bowser and colleagues focused on the cytoplasmic activities of RBM45. They found that like TDP-43 and FUS, it moved from the nucleus to the cytoplasm in cultured motor neurons that were undergoing oxidative stress. There, it sometimes co-localized with stress granule markers, but it also bound KEAP1, a suppressor of the cell’s oxidative stress response. Mislocalized, cytoplasmic RBM45 prevented KEAP1 degradation, diminishing the stress response and leaving neurons vulnerable to attack by free radicals. During ALS, motor neurons experience oxidative stress, so RBM45’s actions could be a reason they fail to protect themselves (Bakkar et al., 2015).
In sum, RBM45 associates with stress-induced structures both in the cytoplasm and in the nucleus of neurons afflicted with neurodegenerative disease, suggesting the nucleus may be a new locus of pathology. “It is another protein that probably plays roles in multiple steps of RNA metabolism and is altered during times of stress and injury,” Bowser told Alzforum. “It opens up new opportunities to explore how nuclear stress is critical for determining long-term cell survival of neurons.”
Peter Ash of Boston University, who did not participate in the studies, said more work is needed to understand the implications of RBM45 in nuclear stress structures. “Perhaps RBM45 is regulating a number of important stress-responsive pathways, such as KEAP1 and HSF1,” he speculated. “Perhaps RBM45 function is key to the sort of appropriate and transient stress response that is lost in ALS.”
Paul Taylor of St. Jude Children’s Research Hospital in Memphis, Tennessee, said the work was important. “There may be a direct link between RBM45 function and disease, although this remains to be firmly established,” commented Taylor, who co-organized the symposium. “The biology of this protein is a strong reminder that the pathobiology of ALS likely involves disturbance of multiple RNA-protein assemblies that reside in both nuclear and cytosolic compartments.”
Amelie Gubitz of the National Institute of Neurological Disorders and Stroke in Bethesda, Maryland, praised the insights derived from a fishing expedition in CSF. “I think this type of patient biosample-based research has great potential in terms of developing ALS biomarkers and obtaining new insights into the molecular mechanisms of the disease,” she told Alzforum. Bowser said he is interested in exploring RBM45’s potential as a biomarker, but has not yet checked how its levels correlate with disease onset or progression.—Amber Dance
ALS Model Mice Roar Back When Human Transgene Silenced
Neurodegeneration is usually pretty permanent—but TDP-43 mice are making a comeback. One line of mice deteriorated during six weeks when a TDP-43 transgene was active, but improved rapidly once it was shut off, regaining the strength to grip a wire and balance on a rotating rod. Virginia Lee of the University of Pennsylvania Perelman School of Medicine in Philadelphia presented these data at RNA Metabolism in Neurological Disease, a satellite meeting held October 15-16 of the Society for Neuroscience annual meeting in Chicago. Lee published some of the findings in the July 22 Acta Neuropathologica online. Also at the meeting, Krista Spiller from Lee’s lab reported that the first motor neurons to die off in these mice match those most vulnerable in humans—the fast-fatigable motor neurons—and that more-resistant neurons can sprout additional axons and take their place once the toxic TDP-43 is gone. In the October 5 issue of the same journal, Lars Ittner and colleagues at the University of New South Wales in Sydney report on an inducible TDP-43 mouse which develops movement and memory problems, but also recovers once the transgene is silenced.
Business as usual.
Turning off TDP43 allowed axons (red) to link back up with motor endplates on the muscle cells (green). [Courtesy of Acta Neuropathologica/Springer]
TDP-43 forms aggregates in most cases of amyotrophic lateral sclerosis, as well as some cases of frontotemporal dementia and Alzheimer’s disease. Lee’s results suggest that treatments aimed at TDP-43 proteinopathy could provide people with some level of recovery, even after neurodegeneration has commenced, said Wilfried Rossoll of Emory University in Atlanta. He did not participate in the study.
TDP-43 proteinopathy has proven difficult to model in mice (see Sep 2012 news). Part of the problem is that the protein, which shuttles between the nucleus and cytosol to manage RNAs, is required for cell viability. Lee and others suspect that in disease states it fails to perform its normal function, but that has been hard to test because knockouts do not survive (see Mar 2010 news). Since pathological TDP-43 aggregates in the cytoplasm, Lee decided to focus on that, deleting the protein’s nuclear localization sequence from the human gene before putting it into a mouse. First author Adam Walker, now at Macquarie University in Sydney, used a neurofilament heavy chain promoter, which is active in all neurons, to drive expression of the transgene. A tetracycline response element ensured the gene would remain inactive and the mice developed normally so long as their chow contained the inhibitor doxycycline.
The researchers dubbed the mice “regulatable NLS” or rNLS. When the mice reached about five weeks old, the researchers switched their diet to dox-free food and watched the disease unfold. The mice developed TDP-43 inclusions in the spinal cord and brain. In addition, the animals’ brains shrank and their motor neurons retracted from muscles and died. Researchers at the meeting found the model intriguing. Compared to other TDP-43 models, the pathology in the rNLS mice appeared more akin to that in people, commented Dieter Edbauer of Ludwig-Maximilians University of Munich, who did not participate in the work.
The animals also experienced a progressive motor neuron disease. They developed tremors in their paws and lost the ability to grip a wire or balance on a rotating rod. They lost weight, and died about 10 weeks after they stopped eating dox-laced chow. Lee thinks the disease resulted from the diminished expression of endogenous, full-length TDP-43, which she said disappeared within a week of the transgene’s activation. Because TDP-43 suppresses its own transcription, the transgene presumably turned off the regular mouse gene, suggested Lee (see Jan 2011 news).This would compromise management of RNA trafficking and translation.
The real excitement happened when Walker and colleagues returned the dox chow to some of the mice that had been dox-free for six weeks. A week later, the animals started to grip a wire more tightly, and they wobbled less on the rotating rod. They gained weight again, and lived out a normal lifespan. TDP-43 aggregates started to disappear within two weeks on dox, and were completely gone after three months. Cortical and motor neurons stopped degenerating. More than that, neurons that remained seemed to take over for those that had died. Six weeks back on dox doubled the percentage of neuromuscular junctions that were innervated by motor neurons (see image above). The researchers surmised that the motor neurons still present were able to take over for the ones that withered while the TDP-43 transgene was overexpressed.
Spiller analyzed this re-innervation in more depth. First, she figured out which motor neurons were most vulnerable to the TDP-43 toxicity, and which persisted. Previously, scientists had reported that fast-fatigable motor neurons, which innervate fast-twitch muscles and mediate quick movements such as jumping and running, were the first to degenerate in people and in ALS mice overexpressing mutant SOD1, another cause of the disease. In contrast, fast-fatigue-resistant motor neurons, which activate quick movements but are slower to tire, and slow motor neurons, which manage activity like standing or strolling, take longer to degenerate (Pun et al., 2006; Kanning et al., 2010). Though there are few markers for these motor-neuron populations, they usually can be identified by the muscles they connect to and their size. Fast-fatigable neurons link up with fast-twitch muscles and are the largest, while the slow motor neurons attach to slow-twitch muscles and are smaller. In the rNLS mice, Spiller observed that the fast-fatigable motor neurons were the ones that disappeared.
With those fast-fatigable neurons gone, how did the mice recover once their TDP-43 expression re-normalized? To label which motor neurons connected to the re-innervated muscles, Spiller injected the muscles with a fluorescent tracer. From this, she confirmed that disease-resistant neurons, the smaller fast-fatigue-resistant and slow ones, had sprouted new connections. These same fatigue-resistant or slow motor neurons also started to express MMP9, a metalloproteinase typically found only in fast-fatigable neurons. Spiller said she has not yet worked out the significance of the new MMP9 production.
“The recovery was really remarkable to see,” Spiller said. “Even after a mouse has lost a third of its motor neurons, it could still recover … and walk around the cage.” That makes targeting TDP-43 seem like a viable therapeutic strategy, she suggested, even for people who have already lost motor neurons. This kind of recovery is encouraging, agreed Paul Taylor of St. Jude Children’s Research Hospital in Memphis, Tennessee, who co-organized the symposium but did not participate in Lee’s research.
Notably, researchers working with two other lines of TDP-43 mice have observed recovery as well. In another line of repressible TDP-43 mice, this time with the CaMKIIa promoter driving expression of a cytosolic construct in the forebrain, have also observed improvements in behavior when they turn off TDP-43. With TDP-43 on, these mice have learning and memory defects and motor problems. Turning it off reversed most of the symptoms within just two weeks, so long as neurodegeneration had not progressed too far (Alfieri et al., 2014). “Both animal models represent complementary tools to address questions regarding the role of altered TDP-43 in ALS-like or FTLD-like phenotypes,” commented that study’s author, Lionel Igaz of the University of Buenos Aires in Argentina (see full comment, below). And Ittner and colleagues generated mice with an inducible TDP-43 transgene including a disease-linked mutation. Overexpression of the transgene in neurons induced neurodegeneration, leading to deficits in movement, spatial memory and disinhibition. Turning off the transgene led to significant improvements in those symptoms in just one week (Ke et al., 2015).—Amber Dance
Stribl C, Samara A, Trümbach D, Peis R, Neumann M, Fuchs H, Gailus-Durner V, Hrabě de Angelis M, Rathkolb B, Wolf E, Beckers J, Horsch M, Neff F, Kremmer E, Koob S, Reichert AS, Hans W, Rozman J, Klingenspor M, Aichler M, Walch AK, Becker L, Klopstock T, Glasl L, Hölter SM, Wurst W, Floss T.
Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43.
J Biol Chem. 2014 Apr 11;289(15):10769-84. Epub 2014 Feb 10
PubMed.
Unexpected Polypeptides Pop Up in Huntington’s Disease
Scientists have been missing out on four repeat polypeptides in Huntington’s disease, according to new data from the lab of Laura Ranum at the University of Florida in Gainesville. She discovered that the same trinucleotide repeats that encode well-known polyglutamine tracts in huntingtin can also be translated, sans start codon, into polyserine, polyalanine, polyleucine, and polycysteine. Moreover, the novel polypeptides appear alongside the polyglutamine-expanded huntingtin, and sometimes even overshadow it, in the parts of the brain that degenerate in HD. Ranum presented these results at RNA Metabolism in Neurological Disease, a satellite symposium to the Society for Neuroscience annual meeting, held October 15-16 in Chicago. Several scientists in attendance called Ranum’s news one of the highlights of the conference. The work also appears in the November 18 Neuron.
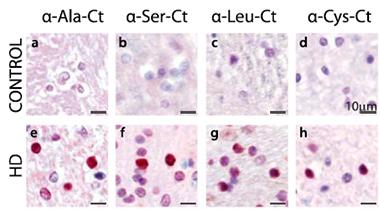
Polypeptide variety.
The striata of people who died of HD, but not controls, contained four RAN polypeptides. [Courtesy of Neuron, Bañez-Coronel et al.]
Previously, Ranum discovered that repetitive gene sequences sometimes get processed through what is known as repeat-associated non-ATG (RAN) translation (Zu et al., 2011). For example, hexanucleotide repeats in the C9ORF72 gene, linked to both amyotrophic lateral sclerosis and frontotemporal dementia, are copied and translated in both the sense and antisense directions, yielding a set of poly-dipeptides that scientists suspect are toxic (see Nov 2013 news). However, those repeats—and every other sequence observed to undergo RAN translation—reside in an intron. Could the same thing happen in coding repeats?
To find out, first author Monica Bañez-Coronel and colleagues picked the huntingtin gene as a possible candidate because it contains a stretch of CAG codons. People with 36 or more of these repeats are at risk for HD, and those with 40 or more are almost certain to develop the disease. Upward of 50 repeats, carriers are likely to develop HD early in life, some even before adulthood. If those CAG codons were to undergo RAN translation in the sense direction, they could produce polyserines and polyalanines, in addition to the canonical polyglutamine. The antisense strand would yield polyleucine, polycysteine, and polyalanine. The authors wanted to generate antibodies to each, but it can be difficult to make antibodies to repetitive peptides, Ranum said.
Fortunately, each potential polypeptide would be followed by a unique amino acid sequence as translation continued beyond the repeats until it hit a stop codon. Bañez-Coronel generated antibodies to four of those post-repeat peptides. (The strategy did not work for polyalanine because the stop codon occurred right after the repeat). She applied these antibodies to autopsy samples taken from the striatum, a brain region usually affected in HD. All four lit up neurons, astrocytes, and microglia in brains from people with Huntington’s, but not controls (see image above). The polypeptides were found in the nucleus and cytoplasm, and appeared diffuse and in punctate aggregates.
Bañez-Coronel compared immunohistochemistry for polyglutamine-expanded huntingtin to that of the RAN-translated poly amino acids in different subregions of the striatum. Notably, in the caudate and putamen, she found more cells positive for RAN polypeptides than for polyglutamine. In the white-matter tracts associated with those regions she saw intense RAN protein signals, whereas polyglutamine did not appear at all.
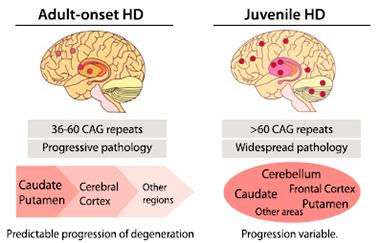
Step by step.
Neurodegeneration progresses predictably in people with adult-onset disease, but varies in those with juvenile-onset disorder. [Courtesy of Neuron, Bañez-Coronel et al.]
The striatal regions of the brain are typically the first to degenerate in HD, followed by the cortex. In the adult tissues, Bañez-Coronel detected RAN polypeptides in the cortex and cerebellum as well, though the antibody signal was less intense than in the striatum. Polyglutamine, in contrast, occurred in the cortex but never reached the cerebellum.
In juvenile-onset cases, progression of pathology varies (see image at left). The authors wondered if RAN polypeptides might help explain that variation. They examined brains from five juvenile cases. All expressed RAN proteins in the striatum, frontal cortex, and cerebellum. In the two brains with the most severe cerebellar atrophy, RAN peptides, but not polyglutamine, appeared in the cerebellum, Ranum said.
In addition to its variable sites of pathology, juvenile HD typically results from longer expansions in the Htt gene. To find out how repeat length affects RAN translation, the authors transfected HEK293T cells with minigenes containing huntingtin’s first exon and 23-80 repeats. They focused on the sense polypeptides containing alanine or serine, since they were comparing them to the polyglutamine-containing huntingtin fragment produced by normal, ATG-associated translation in the sense direction. These minigenes always produced polyglutamine-htt regardless of repeat size. They made polyserine if the repeat length was 35 or more, and polyalanine when the extension grew to 50. In addition, the serine polypeptide started to aggregate once it reached about 45 amino acids long. Thus, the polyalanine and insoluble polyserine associated with longer repeat lengths.
Dieter Edbauer of Ludwig-Maximilians University of Munich, who did not participate in the work, told Alzforum he was particularly intrigued by the results with juvenile HD. Carriers of longer repeats might get sick because of more RAN translation, rather than simply a longer polyglutamine stretch within the huntingtin protein, he said. And the anatomical distribution of those RAN polypeptides differs from the polyglutamine-expanded huntingtin, potentially explaining why different regions are affected in juvenile and adult-onset HD.
Supporting this theory, the authors saw indicators that the RAN polypeptides could be toxic. The brain regions where RAN products were prevalent, the caudate and the putamen and their white-matter bundles, tested positive for Iba1, a microglial marker of neuroinflammation. Those spots also tested positive for caspase 3, a marker of apoptosis, indicating that those neurons and glia with RAN polypeptides were sick or dying. Bañez-Coronel confirmed that RAN polypeptides were toxic when she transfected minigenes into neuronal and glial cell cultures.
“These new mutant proteins may contribute to the disease,” Ranum concluded. At this point, she added, she cannot be sure about the relevant contributions of polyglutamine versus the RAN polypeptides. One implication, the authors noted, is that researchers trying to treat Huntington’s may have to expand their ideas to interfere with RAN translation in both the sense and antisense directions.
“The finding of intense RAN protein accumulation in white-matter regions in the absence of significant polyglutamine accumulation is particularly interesting,” Bañez-Coronel told Alzforum. “White-matter changes can occur early in disease and are also detected in presymptomatic patients. Our findings suggest that RAN proteins are likely contributing to these white-matter alterations.”
In addition, Ranum pointed out that several other neurological diseases, including spinocerebellar ataxia and spinal-bulbar muscular atrophy, are associated with polyglutamine stretches in coding regions. She speculated that RAN translation might occur in those conditions as well. Tom Cooper of the Baylor College of Medicine, Houston, told Alzforum that Ranum’s work convinced him to be on the lookout for RAN translation. He will be checking his mice modeling myotonic dystrophy type 1 for signs of RAN polypeptides.—Amber Dance
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP.
Non-ATG-initiated translation directed by microsatellite expansions.
Proc Natl Acad Sci U S A. 2011 Jan 4;108(1):260-5. Epub 2010 Dec 20
PubMed.







Comments
No Available Comments
Make a Comment
To make a comment you must login or register.