C9ORF72 Mice Point to Gain of Toxic Function in ALS, FTD
Quick Links
Over the past several years, much research has pointed to cellular mishandling of RNA as a key factor in neurodegeneration. Numerous RNA-binding proteins—from the well-known TDP-43 and fragile X mental retardation proteins to relative newcomers such as hnRNPA3 and RBM45—mutate or aggregate in diseases such as amyotrophic lateral sclerosis, frontotemporal dementia, and myotonic dystrophy. As a result, scientists believe RNAs may be misspliced, sequestered from the ribosome, or otherwise fouled up. This convergence of mechanisms made the RNA Metabolism in Neurological Disease conference, held October 15-16 in Chicago before the Society for Neuroscience annual meeting, the place to be for scientists interested in ALS, FTD, and similar disorders. Attendees were astounded by the latest news about RNA-binding proteins and C9ORF72, a gene with ALS- and FTD-linked repeat expansions that encode anomalous repetitive RNAs and peptides. Co-organized by Paul Taylor of St. Jude Children’s Research Hospital in Memphis, Tennessee, and Fen-Biao Gao of the University of Massachusetts Medical School in Worcester, the meeting featured new mouse models of C9ORF72 disease. Scientists who eliminated the gene from their animals noticed powerful inflammation, while those who expressed a mutant transgene with repeats detected cognitive and motor defects.
C9ORF72 Mouse Models
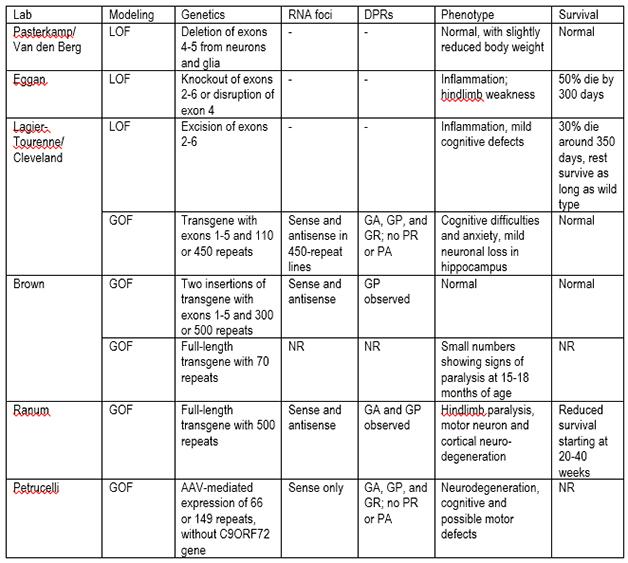
C9ORF72 model mice. LOF=loss of function; GOF=gain of function; DPRs=dipeptide repeats; NR=not reported; GA=glycine-alanine; GP=glycine, proline; GR=glycine-arginine; PR=proline-arginine; PA=proline-alanine.
C9ORF72 encodes a protein of uncertain function. Hexanucleotide repeats in the gene’s first intron—numbering in the hundreds or thousands—have been linked to ALS and FTD (see Sep 2011 news). Cells transcribe these repeats into messenger RNAs in both the sense and antisense directions, and these RNAs aggregate into nuclear foci (see Jan 2013 conference news; Nov 2013 news). Despite being in introns, the repeats somehow avoid getting spliced out of mRNAs—scientists are not yet sure how—and ribosomes then translate them through a process known as repeat-associated non-ATG translation. Five different repeat dipeptides result from the six possible reading frames in the sense and antisense directions (two reading frames encode the same dipeptides). These peptides can gather into toxic aggregates (see Feb 2013 news).
Researchers are trying to work out how the repeats instigate disease by differentiating among three hypotheses. For one, the repeats interfere with normal C9ORF72 production, leading to a loss of function when people only make half the normal amount of the protein. Two, the RNA foci might cause a gain of function that damages neurons. Three, the translated dipeptides might be toxic. Several research groups attempting to model these scenarios in mice presented their preliminary results in Chicago (see table above). The upshot: No model quite mimics the human condition, said Robert Brown of the University of Massachusetts Medical School in Worcester, who has engineered two transgenic lines. With multiple mouse lines still under study, it is too soon to draw firm conclusions about ALS and FTD from the animals, said Clotilde Lagier-Tourenne of Massachusetts General Hospital in Charlestown, who is analyzing both loss-of-function and gain-of-function models. Nonetheless, researchers at the meeting seemed confident that these mice will offer clues about pathology. They will also serve as testing grounds for therapeutics, such as antisense oligonucleotide therapy to eliminate repeat RNAs and their protein products. “We are making lots of progress building models for C9ORF72,” said Lagier-Tourenne. “It is very exciting.”
Knocking Out C9ORF72 Kicks Up Cytokine Storm
Two groups addressed the loss-of-function idea by generating C9ORF72 knockout mice. Kevin Eggan and colleagues at Harvard University, and a collaboration between Lagier-Tourenne’s group and Don Cleveland’s lab at the University of California, San Diego, generated mice from the same embryonic stem cell line that lacks exons 2–6 of the 12 found in the endogenous C9ORF72 gene. Mice heterozygous for the truncated gene made 50 percent of the normal amount of C9ORF72 RNA and protein. Homozygous mice completely lacked both transcript and protein.
The homozygote knockouts exhibited problems reminiscent of ALS and FTD. Eggan said their animals developed weakness in their hind legs and struggled to breathe as they aged. Lagier-Tourenne said their mice had no obvious motor defects but developed mild behavioral abnormalities at six months. They erred navigating mazes and the males were highly anxious, said Jie Jiang of UC San Diego, who presented the data on a poster.
Eggan’s group used electromyography to detect denervation at the neuromuscular junction in their animals, but came up with baffling results. They found denervation and reduced motor neuron counts in the first group of animals they assessed, but not in the next, supposedly identical cohort. He has not yet figured out why. The knockouts died early. Jiang reported that about 30 percent of his mice died after around a year, while less than half of Eggan’s mice made it to their first birthday.
While the Cleveland/Lagier-Tourenne heterozygotes survived normally, the Eggan heterozygotes exhibited a diluted version of the homozygous phenotype, and some of them died within a year as well. The differences between the populations might be simply due to the animals’ environments, Jiang said, noting that mice are quite sensitive and even something like construction noise outside could impact their phenotype.
Eggan noted that the mice died from a variety of causes. Some suffered respiratory failure. Others exhibited such severe loss of motor control or skin problems that veterinarians recommended they be sacrificed. Some were found, on necropsy, to have bled internally. The most striking change, Eggan said, was grossly enlarged spleens in the homozygous C9ORF72 knockouts (see image below). Many mice also had swollen lymph nodes in their necks. Lagier-Tourenne and colleagues noticed enlarged spleens and lymph nodes in their animals, as well.

Immune indicator
C9ORF72 knockout mice possessed much larger spleens (right) than normal mice (left), suggesting inflammation. [Image courtesy of Aaron Burberrry, Harvard University.]
The enlarged organs indicated defects in the body’s blood-cell supply, which Eggan’s group pursued further. The animals produced slightly lower red blood cell counts and fewer platelets than normal, but an excess of white blood cells. These findings made the researchers suspect an imbalance in cytokines, immune signaling molecules. Indeed, numerous cytokines, including interferon-γ and IL-5 were upregulated, mimicking a hyper-immune reaction sometimes termed a “cytokine storm,” which can be fatal in humans. The blood and cerebrospinal fluid of people with ALS also contains upregulated cytokines—IL-17 and IL-23—Eggan noted, though he said the cause may be different (Rentzos et al., 2010).
To determine if the mouse defects resulted from C9ORF72 loss in the hematopoietic system, Eggan’s group transplanted bone marrow from C9ORF72 knockouts into wild-type mice and vice versa. One of the first things they noticed was that wild-type animals, upon receiving knockout bone marrow, developed bulky chests due to swollen lymph nodes beneath their skin. Their spleens grew large, though not as large as those from full C9ORF72 knockouts. Their lifespan was shortened, though not as much as complete knockout mice. Transplanting wild-type bone marrow into knockout mice partially rescued the enlarged spleen phenotype, and enhanced survival.
Eggan’s group used CRISPR gene editing to generate a separate line of mice (see Sep 2014 series) with a premature stop codon in exon 4. The researchers have not yet fully evaluated the RNA and protein levels in these mice, but already know that they, too, have anemia, cytokine storm, and enlarged spleens. As with the other knockout lines, many died before a year of age.
The signs are not reminiscent of neurodegeneration but of blood cancer, commented Jeffrey Rothstein of Johns Hopkins University in Baltimore. Lagier-Tourenne said pathologists told her the mice appeared to have lymphoma. However, the cancer experts Eggan consulted told him he was seeing something else. “It’s more of a massive inflammatory condition than cancer,” he said. He speculated that inflammation might be behind some motor deficits of the knockouts, such as falling off a rotating rod. He quipped that he, too, might drop off a rotating rod if he were weathering a cytokine storm.
Dieter Edbauer of Ludwig-Maximilians University of Munich agreed that these mice indicate that C9ORF72 performs some function in the immune system. However, he, Jiang, and Eggan pointed out that knockout mice with no C9ORF72 are not directly comparable to people with the C9ORF72 repeat expansion because the people make some C9ORF72 protein from their one good gene. At least, Jiang said, the mice allow him to conclude that loss of C9ORF72 function by itself does not precipitate neurodegeneration. In this he agrees with the authors of a recent paper who observed no signs of neurodegeneration or change to mortality in mice missing C9ORF72 only in their nervous system. Those mice had normal levels of the gene in the rest of the body, which presumably precluded the inflammatory blood phenotype (see Jun 2015 news).
Wrangling Repeats into Mice Yields Conflicting Results
What, then, happens to transgenic mice modeling gain of function? The first published gain-of-function model, discussed by Leonard Petrucelli of the Mayo Clinic in Jacksonville, Florida, relies on AAV-mediated expression of only the hexanucleotide repeat in the brain and spinal cord. These mice, expressing 66 repeat copies, mimic several characteristics of human disease: RNA foci and dipeptide inclusions as well as TDP-43 aggregates in the brain, plus motor and cognitive symptoms (see May 2015 news). Petrucelli’s lab is now working on a new version with 149 repeats, with which they have already detected poly-glycine-proline dipeptides, he said.
Other scientists created traditional transgenic mice, starting with human C9ORF72 genes copied into bacterial artificial chromosomes (BACs), then dropped into the mouse genome. Both the Brown lab and the Cleveland/Lagier-Tourenne collaboration started with the same partial C9ORF72 construct, comprising the gene’s first five exons and including a 450 hexanucleotide expansion. However, because the guanine- and cytosine-rich repeats are unstable, the expansion can grow or shrink in bacterial hosts. The upshot was that the mice had different repeat numbers, though once in a mouse the given sequence length stays stable, Jiang noted.
He and Lagier-Tourenne said they managed to create several mouse lines, selecting four for detailed analysis. One line carried a transgene with 110 repeats, and three contained a gene with 450 repeats though expressed at different levels. This panel, including both homozygous and heterozygous BACs, allowed the researchers to measure pathology at varying doses of the transgene. The line they have best characterized has 450 repeats and the transgene is expressed at about 3.5 times endogenous C9ORF72 levels, Jiang told Alzforum. Meanwhile, Brown’s lab obtained one stable mouse line with two transgene insertions in the genome, one with 300 repeats and the other with 500, yielding four total transgenes in the homozygotes. The researchers analyzed animals with transgene expression similar to that of the endogenous gene, Brown said.
Brown and colleagues observed a slight decrease in motor neuron numbers compared to control mice, but it was not statistically significant, he said. The scientists saw no evidence of denervation at the neuromuscular junction. Lagier-Tourenne reported that the lines with 450 repeats, at 12 months, exhibited mild loss of hippocampal neurons.
As suspected, the C9ORF72 pathology was dose-dependent, in terms of both repeat number and expression level. RNA foci, both sense and antisense, appeared in mice with 300 to 500 repeats, but not mice with only 110 repeats. Animals with greater transgene expression exhibited more foci as well. Cleveland and Lagier-Tourenne also detected the three dipeptide repeats translated from the sense C9ORF72 RNA, poly-glycine-alanine, poly-glycine-proline, and poly-glycine-arginine. Using an immunoassay specific for glycine-proline, both groups detected soluble dipeptide repeats in the brains of young mice. The soluble peptide transitioned to immunoreactive aggregates in older animals, Lagier-Tourenne said.
Though the mice exhibited these molecular signs of C9ORF72 disease, there was little wrong with them physically. The Lagier-Tourenne/Cleveland mice moved normally, though by one year of age they struggled to navigate mazes, and exhibited mild neurodegeneration in the hippocampus. They lived normal lifespans. Brown’s animals, now two years old, are of normal weight and without motor defects. They were also cognitively normal in behavioral tests including marble burying—anxious mice entomb the unfamiliar objects—and dominance interactions between old and young mice, Brown reported. Those animals carried just the first five exons of the C9ORF72 gene.
In addition, Brown’s group generated a mouse line that contains the full-length C9ORF72 transgene, albeit with only 70 repeats. The researchers have not analyzed the animals thoroughly yet, but for now Brown reported that some of them are starting to exhibit paralysis at 15 to18 months of age.
Laura Ranum of the University of Florida in Gainesville, also generated mice that contain full-length C9ORF72 transgene, with 500 repeats. Some of the mice died between 20 and 40 weeks of age, their demise preceded by weight loss, failing grip strength, and inability to move their hind legs. Others survived longer, but by one year, 80 percent had died or developed phenotypes such as anxiety and a hunched back typical of some other ALS models. The scientists observed neuron loss consistent with both ALS, with fewer motor neurons in the spinal cord and denervation at neuromuscular junctions, and FTD, with neurodegeneration in the cortex. The animals also accumulated sense and antisense RNA foci in their brains and spinal cords, as well as aggregated dipeptide repeats.
“All these mice suggest that the repeats have a gain–of-function toxicity,” Jiang concluded. However, researchers were left scratching their heads about the variation between the lines. The data so far seem to indicate that the full-length C9ORF72 transgene might somehow be more toxic than the partial construct, but researchers need to compare notes, Brown said. Lagier-Tourenne cautioned that scientists check the sequences of their constructs carefully. She speculated that extra genes contained in the short or long BAC clones, including possibly unannotated ones, might be responsible for some phenotypes. Taylor also theorized that something in the larger transgene might influence transcription levels for the antisense RNAs. “We are not at the end of the road yet,” he said.
Indeed, Lagier-Tourenne and Cleveland are already plotting their next mouse line, a cross between their heterozygous C9ORF72 knockouts and the transgenics with the repeats. This, they hope, will yield an animal more representative of human C9ORF72 expansion carriers, who have half the normal complement of the normal gene along with the repeat-expanded version.—Amber Dance
References
News Citations
- Corrupt Code: DNA Repeats Are Common Cause for ALS and FTD
- Chicago—RNA Inclusions Offer Therapeutic Target in ALS
- Sense, Antisense: C9ORF72 Makes Both Forms of RNA, Peptides
- RNA Twist: C9ORF72 Intron Expansion Makes Aggregating Protein
- No C9ORF72, No Problem: Knockout Mouse Neurologically OK
- First C9ORF72 Mice Mimic Key Pathology, Behavior
Series Citations
Paper Citations
- Rentzos M, Rombos A, Nikolaou C, Zoga M, Zouvelou V, Dimitrakopoulos A, Alexakis T, Tsoutsou A, Samakovli A, Michalopoulou M, Evdokimidis J. Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation?. Acta Neurol Scand. 2010 Dec;122(6):425-9. PubMed.
Further Reading
Papers
- Harms MB, Cady J, Zaidman C, Cooper P, Bali T, Allred P, Cruchaga C, Baughn M, Libby RT, Pestronk A, Goate A, Ravits J, Baloh RH. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging. 2013 Sep;34(9):2234.e13-9. PubMed.
- Cooper-Knock J, Kirby J, Highley R, Shaw PJ. The Spectrum of C9orf72-mediated Neurodegeneration and Amyotrophic Lateral Sclerosis. Neurotherapeutics. 2015 Apr;12(2):326-39. PubMed.
- Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S, Sidle K, Fratta P, Orrell RW, Hardy J, Holton J, Revesz T, Rossor MN, Warren JD. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol. 2015 Mar;14(3):291-301. Epub 2015 Jan 29 PubMed.
- Philips T, Rothstein JD. Rodent Models of Amyotrophic Lateral Sclerosis. Curr Protoc Pharmacol. 2015 Jun 1;69:5.67.1-5.67.21. PubMed.
News
- Up-and-Coming ALS Mice Leave Scientists ConFUSed
- DC: Myriad Mice Mimic ALS, FTLD, Loss of TDP-43 Function
- Going Wild About the Latest TDP-43 Mouse Models
- DC: New ALS Genetics Hog the Limelight at Satellite Conference
- C9ORF72 Steals the Show at Frontotemporal Dementia Meeting
- Researchers Revel in C9ORF72 Advances at RNA Symposium
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.