Surprise Save: Excitability Protects Neurons from Lou Gehrig’s
Quick Links
Some motor neurons succumb to the pathology of amyotrophic lateral sclerosis early in the disease, while others manage to survive much longer. In the October 2 Neuron, researchers suggest that the most vulnerable neurons are those that are least electrically excitable, while the more resistant ones are more easily excited. Boosting excitability protected motor neurons, the research shows. The findings appear counterintuitive, because an established hypothesis holds that in ALS, aka Lou Gehrig's disease, neural excitation produces excess glutamate that in turn damages motor neurons. The scientists speculated that when and how strongly excitation occurs might determine whether it benefits or harms the cell. It is possible that early in the disease excitation protects neurons, while later on too much of it is toxic, they suggested.
Senior author Pico Caroni and colleagues at the Friedrich Miescher Institute in Basel, Switzerland, study motor neurons’ vulnerability to mutant superoxide dismutase 1 (SOD1), a major cause of familial ALS (see ARF related news story on Saxena et al., 2009). Neurons fall victim to this disease in three waves. First to falter are the large, rapid-firing, and fast-fatigable motor neurons that mediate quick actions, such as jumping. Next to go are the rapid-firing but fast-fatigue-resistant motor neurons, which also generate quick movements but tire more slowly. Finally, the slow motor neurons that control extended activities, such as standing, begin to degenerate (Pun et al., 2006). Researchers know little about the first group other than that they are less excitable than other types, Caroni told Alzforum. In the current work, joint first authors Smita Saxena and Francesco Roselli investigated whether excitability influences vulnerability. Saxena has since moved to the University of Bern, Switzerland.
The Benefits of Excitation
First, the researchers needed a method to assess motor neuron pathology early on in disease. They used antibodies against misfolded SOD1 (Gros-Louis et al., 2010) to time its appearance in fast-fatigable and fast-fatigue-resistant motor neurons in mice overexpressing mutant human SOD1. They saw that misfolded SOD1 appeared in the first week of life, well before symptoms, in the fast-fatigable cells, but only arose just before symptom onset in the fatigue-resistant neurons. This pattern emerged in a rapid-disease model that accumulates SOD1 beginning at 2 months of age, and in a more slowly progressing model that shows signs of pathology at 5 months.
Next, Saxena and colleagues asked whether altering the excitability of particular neuron types would change the buildup of misfolded SOD1. They expressed either of two chimeric ion channels in the motor neurons as tools to render the cells more excitable or less. These chimeric channels had been previously engineered such that they could be activated with small molecules (Magnus et al., 2011). Both versions include cation channels coupled to a portion of the acetylcholine receptor, and turn on when that receptor portion binds a benzamide ligand. One version includes part of a serotonin receptor and, once its ligand has bound, depolarizes neurons, enhancing their excitability. The other includes a portion of a glycine receptor, and ligand binding causes hyperpolarization and reduced excitability of the neuron.
To express these channels specifically in motor neurons, Saxena and colleagues used a Cre-Lox system with Cre activated by the choline acetyltransferase promoter, which is active in motor neurons. They targeted the floxed ion channel genes to one side of the lumbar spinal cord with a viral vector, leaving the other side to act as an internal control. They then injected receptor ligands daily, to activate these engineered channels and manipulate neuronal excitability at will.
Boosting motor neuron excitability with the serotonin chimera reduced SOD1 misfolding. Dampening excitability increased misfolding. In fact, the SOD1 aggregates in the hyperexcitable neurons seemed to disappear, Caroni noted, suggesting that it is possible to dissolve them (see image below). The treatment also delayed denervation of the muscles innervated by the fast-fatigable motor neurons.
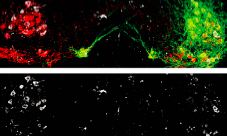
Motor neurons (red) expressing an activating channel (green) exhibited fewer SOD1 aggregates (lower panel) than motor neurons without the channel on the contralateral side of the spinal cord (left side of both panels) Image courtesy Pico Caroni and Neuron, October 2, 2013
The researchers theorized that the intensified excitability might be a defense mechanism. Going back to the regular mutant SOD1 model, they noticed that motor neurons had fewer inhibitory synapses and more excitatory synapses than did the same neurons in control, wild-type animals. Caroni believes this to be a natural neuroprotective response to the accumulated SOD1. In particular, cholinergic C-boutons, a type of synapse that promotes excitation, were larger in SOD1 animals than in controls.
Molecular Mechanisms
Does a neuron’s excitability influence how long it survives? To answer that, Saxena and her colleagues focused on the C-boutons, reasoning that if they promote excitability and neuroprotection, then blocking input to them should speed up pathology. Sure enough, treating the mice with methoctramine, a commercial muscarinic receptor agonist that specifically blocks acetylcholine signaling to C-boutons, drove up levels of misfolded SOD1 in motor neurons.
Next, the scientists examined how C-bouton signaling might protect neurons by comparing expression and activation of several candidate molecules in vulnerable and resistant neurons in mSOD1 mice. They noticed that the receptor mammalian target of rapamycin, aka mTOR, was highly activated in the vulnerable motor neurons. Treating the mice with the mTOR inhibitor rapamycin increased SOD1 misfolding further, suggesting that mTOR activity protects the neurons. The researchers do not know how the C-boutons activate mTOR, or how mTOR might reduce SOD1 misfolding.
In a pilot study, five SOD1 mice treated with rapamycin died about a month sooner than did five controls, supporting the idea that mTOR signaling is protective. The detrimental effects of rapamycin here stand in direct contrast to the protective effects seen with this drug in other neurodegenerative diseases, including Parkinson’s and Huntington’s, noted Mark Mattson of the National Institute on Aging in Baltimore, Maryland, in a preview accompanying the Neuron study (see Zhang et al., 2011; Sarkar et al., 2009). Rapamycin is an immunosuppressant drug used in organ transplantation.
The mTOR protein participates in numerous pathways where it could contribute to disease. The finding in the current study should remind scientists that they cannot always generalize between conditions, said Christopher Henderson of Columbia University in New York, who was not involved in the study. Other commentators noted that they would like to see the experiment repeated with larger numbers of animals. Typically, ALS researchers prefer to use at least 20 SOD1 animals per treatment group because their survival and behavioral phenotypes are variable, said Brett Morrison of Johns Hopkins University in Baltimore, who was not involved with the work (see Shineman et al., 2011). While a small handful of animals would be sufficient for the majority of biochemical and staining experiments in the paper, the survival results are more provocative than conclusive, Morrison said.
Too Much of a Good Thing?
Overall, Caroni’s results point to excitability as a neuroprotective response to the mutant SOD1 gene. This conclusion surprised Mattson, who noted in his preview that several other studies suggest that motor neurons die, in part, due to the toxicity of excess glutamate produced by over-firing (reviewed in Rothstein, 2009; and see Alzforum news story on Cucchiaroni et al., 2010). Notably, riluzole, the only drug clinically approved for ALS, is thought to work by diminishing motor neuron excitation. “Most people thought that hyper-excitability is bad, and leads to degeneration later on,” said Henderson.
In an effort to reconcile Caroni’s results with previous work, Mattson suggested the phenomenon of hormesis, in which low levels of a stimulus are beneficial, while higher exposure becomes detrimental. In addition, timing could play a role, suggested Morrison. That is, low-level excitability in presymptomatic stages could stave off disease for a while, but excess excitation in the final stages—when riluzole might work best—could hasten it. Whether the same phenomenon would hold true in cases of ALS due to mutations in genes other than SOD1, or sporadic cases, remains to be determined, Mattson wrote.
How about protective excitability in people? Exercise, which forces motor neurons to fire more often, protects some kinds of neurons, Mattson noted (reviewed in Stranahan and Mattson, 2012). However, some studies (for example, Scarmeas et al., 2002) suggest that elite athletes are at higher-than-average risk for ALS. One cannot necessarily equate exercise with neural excitability, Henderson pointed out. Little is known about excitability in the motor neurons of people with ALS, and experts who spoke with Alzforum could not immediately suggest ways to study it.
With regard to therapies modulating neural excitability, clinicians recently halted the Phase 3 trial of ceftriaxone, a drug that dampens excitotoxicity, because it showed no improvement over placebo (see Alzforum news story).—Amber Dance
References
News Citations
- ER Struggles in Motor Neurons That Fall to ALS
- Glutamate Gums Up Motor, Dopaminergic Neurons
- Chicago—ALS Clinical Trials: New Hope After Phase 3 Setbacks
Paper Citations
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009 May;12(5):627-36. PubMed.
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006 Mar;9(3):408-19. PubMed.
- Gros-Louis F, Soucy G, Larivière R, Julien JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem. 2010 Jun;113(5):1188-99. PubMed.
- Magnus CJ, Lee PH, Atasoy D, Su HH, Looger LL, Sternson SM. Chemical and genetic engineering of selective ion channel-ligand interactions. Science. 2011 Sep 2;333(6047):1292-6. PubMed.
- Zhang X, Li L, Chen S, Yang D, Wang Y, Wang Z, Le W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011 Apr;7(4):412-25. PubMed.
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2008 Jul 18; PubMed.
- Shineman DW, Basi GS, Bizon JL, Colton CA, Greenberg BD, Hollister BA, Lincecum J, Leblanc GG, Lee LB, Luo F, Morgan D, Morse I, Refolo LM, Riddell DR, Scearce-Levie K, Sweeney P, Yrjänheikki J, Fillit HM. Accelerating drug discovery for Alzheimer's disease: best practices for preclinical animal studies. Alzheimers Res Ther. 2011;3(5):28. PubMed.
- Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009 Jan;65 Suppl 1:S3-9. PubMed.
- Cucchiaroni ML, Viscomi MT, Bernardi G, Molinari M, Guatteo E, Mercuri NB. Metabotropic glutamate receptor 1 mediates the electrophysiological and toxic actions of the cycad derivative beta-N-Methylamino-L-alanine on substantia nigra pars compacta DAergic neurons. J Neurosci. 2010 Apr 14;30(15):5176-88. PubMed.
- Stranahan AM, Mattson MP. Recruiting adaptive cellular stress responses for successful brain ageing. Nat Rev Neurosci. 2012 Mar;13(3):209-16. PubMed.
- Scarmeas N, Shih T, Stern Y, Ottman R, Rowland LP. Premorbid weight, body mass, and varsity athletics in ALS. Neurology. 2002 Sep 10;59(5):773-5. PubMed.
External Citations
Further Reading
Papers
- Maragakis NJ, Dykes-Hoberg M, Rothstein JD. Altered expression of the glutamate transporter EAAT2b in neurological disease. Ann Neurol. 2004 Apr;55(4):469-77. PubMed.
- Chang PK, Verbich D, McKinney RA. AMPA receptors as drug targets in neurological disease--advantages, caveats, and future outlook. Eur J Neurosci. 2012 Jun;35(12):1908-16. PubMed.
- Lee S, Kim Y, Li E, Park S. Ghrelin protects spinal cord motoneurons against chronic glutamate excitotoxicity by inhibiting microglial activation. Korean J Physiol Pharmacol. 2012 Feb;16(1):43-8. PubMed.
- Aso E, Ferrer I. It may be possible to delay the onset of neurodegenerative diseases with an immunosuppressive drug (rapamycin). Expert Opin Biol Ther. 2013 Sep;13(9):1215-9. PubMed.
- Chong ZZ, Shang YC, Wang S, Maiese K. Shedding new light on neurodegenerative diseases through the mammalian target of rapamycin. Prog Neurobiol. 2012 Nov;99(2):128-48. PubMed.
Primary Papers
- Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P. Neuroprotection through Excitability and mTOR Required in ALS Motoneurons to Delay Disease and Extend Survival. Neuron. 2013 Oct 2;80(1):80-96. PubMed.
- Mattson MP. Excitation BolsTORs Motor Neurons in ALS Mice. Neuron. 2013 Oct 2;80(1):1-3. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Arkansas for Medical Sciences
This confirms a hypothesis that I invoked to explain the results of a study in which I was involved, that examined the role of D-serine in ALS. D-Ser is a co-agonist at the NMDA-R "glycine" site, and is likely to be the most important agonist in the forebrain. However, some evidence had suggested that its overproduction might contribute to excitotoxicity in some instances. John Crow and I crossed the G93A SOD1 mutant mouse with one that lacked the ability to produce D-Ser (though some is still absorbed from food). Expecting the D-Ser reduction to protect from the excitotoxicity that we assumed was contributing to this ALS model, we were surprised to find that the onset of disease was instead hastened. Disease progression was slowed, however, suggesting that excitotoxicity may contribute to pathology in some stages of the disease or in specific neuron populations throughout the course of illness.
References:
Thompson M, Marecki JC, Marinesco S, Labrie V, Roder JC, Barger SW, Crow JP. Paradoxical roles of serine racemase and d-serine in the G93A mSOD1 mouse model of amyotrophic lateral sclerosis. J Neurochem. 2012 Feb;120(4):598-610. PubMed.
Make a Comment
To make a comment you must login or register.