Last month in Madrid, researchers traded news about clinical trials for Alzheimer’s disease. The field is watching the rollout of lecanemab and donanemab with bated breath, debating administration, appropriate use, and hoping for no more deaths. Soon after, European regulators issued a thumbs up for lecanemab. Meanwhile, brain shuttle versions are on the horizon, led by trontinemab. Tau as a target is starting to yield to antibodies, though OGA inhibition currently appears too toxic.
Trontinemab Data Strengthen Hope for Brain Shuttles
Fewer than 1 percent of amyloid-targeted monoclonal antibodies like lecanemab and donanemab reach their targets in the brain. The excess doses required to make up for this problem raise the risk of hazardous brain bleeding, reflected in high rates of amyloid-related imaging abnormalities (ARIA). While the drugs have been approved in the U.S. and some other countries, regulators in Europe have balked, partly due to their risk/benefit ratio. How to improve this? Enter transferrin receptor-based systems that usher antibodies into the brain through its capillaries, where they might avoid the bulk of vascular amyloid thought to provoke ARIA. A handful of these BBB-penetrating candidates are being developed, but so far only one—Roche’s trontinemab—has made its way into clinical trials.
At the Clinical Trial on Alzheimer’s Disease conference, held October 29 to November 1 in Madrid, Roche presented further data from an ongoing dose-finding study, reporting only three mild, asymptomatic ARIA cases so far. At the two highest doses tested, the antibody rapidly vanquished Aβ plaques. The good news was tempered by a death from a cerebral macrohemorrhage after the second dose. MRI scans revealed superficial siderosis and probable cerebral amyloid angiopathy (CAA) at baseline. In response, Roche added these conditions to its exclusion criteria for ongoing and future trials.
Despite this, scientists at CTAD broadly expressed enthusiasm that trontinemab and other TfR-based therapies will pave the way to deliver these antibodies, and other drugs, more safely and efficiently into the brain.
In her keynote speech, Catherine Mummery of University College London called the trontinemab data “fantastically exciting.” She views such strategies as a promising way to make better monoclonal antibodies. “Using active transport mechanisms to get these drugs across the blood-brain barrier will dramatically accelerate the levels of amyloid removal, and equally importantly, really reduce those rates of ARIA,” Mummery said.
Essentially a version of gantenerumab equipped with a so-called Brain Shuttle, trontinemab is an anti-Aβ IgG1 antibody with full effector function and with a TfR-specific Fab fragment attached to one of its Fc domains (image below). In healthy volunteers, the shuttle boosted CNS exposure by eightfold relative to gantenerumab (Mar 2021 conference news).
Tront Transfer. Trontinemab features an anti-Aβ antibody with an anti-TfR “Brain Shuttle” attached (left). Binding to TfR on brain endothelial cells ushers it into the brain via transcytosis (middle). It then engages plaques in the parenchyma, where it exerts full effector function (right). [Courtesy of Luka Kulic, Roche.]
Roche subsequently started dose-finding studies in volunteers with mild cognitive impairment and mild to moderate AD, from which it has been dropping nuggets of data at conferences. Its ongoing Phase 1b/2a study, called Brain Shuttle AD, initially enrolled 60 people split equally among four ascending-dose groups. They received 0.2 mg, 0.6 mg, 1.8 mg, or 3.6 mg trontinemab, or placebo, every four weeks for 28 weeks. At last year’s CTAD, Roche’s Luka Kulic had shown six-month findings from the first three doses and, earlier this year at AD/PD, added preliminary three-month findings from the highest-dose group (Nov 2023 conference news; Mar 2024 conference news). At that time, Kulik said both the 1.8 mg and 3.6 mg doses dramatically reduced amyloid plaques by three months. In the 1.8 mg group, one person developed ARIA-E, another ARIA-H. Among the eight people who had received the 3.6 mg dose for three months, none had yet developed ARIA.
In Madrid, Kulic presented 28-week data from the now-completed part of the trial. He also presented preliminary findings from two additional cohorts, which were added to gather more safety data on the two highest doses. These dose-expansion cohorts will enroll an additional 60 participants each, who will receive either the 1.8 mg or 3.6 mg dose, or placebo, every four weeks for 28 weeks. This second phase is ongoing and, as of the September 2 interim analysis, 60 participants had enrolled in the 1.8 mg group and 40 in the 3.6 mg group. The two groups had received an average of 5.4 and 2.9 doses, respectively.
Kulic first showed safety data from 60 participants who finished Part 1, combined with 100 participants so far enrolled in the ongoing Part 2. For Part 1, Kulic had no new safety issues to report beyond what had been presented at AD/PD, namely that some people reacted to the infusions. This was mitigated in the highest-dose group through pretreatment with anti-inflammatory drugs. Among the 100 participants enrolled in Part 2, Kulic reported two serious adverse events. One was an infusion-related reaction that occurred on a Friday afternoon, prompting clinicians to transfer the participant to a hospital. Kulic said this participant had a fever that quickly subsided.
Alas, the other case proved fatal. Three weeks after receiving her second 1.8 mg dose of trontinemab, a 78-year-old woman developed a right frontal lobar macrohemorrhage. She died two days later. Looking back at her initial screening MRI, the scientists spotted superficial siderosis in her occipital cortex, as well as additional lesions, signifying probable CAA. Amyloid PET scans indicated substantial Aβ accumulation in the same region. She also carried an ApoE2 allele, which is a risk factor for CAA, Kulic said.
The protocol for ongoing and future trontinemab trials has since been amended to exclude people with superficial siderosis. This is in line with published appropriate use recommendations for lecanemab, as well as those for donanemab presented at CTAD (Cummings et al., 2023; see next story).
Warning Sign. Screening MRI from a participant who later died from a cerebral hemorrhage reveal superficial siderosis (top left, arrowheads), as well as other lesions (top right). Substantial Aβ accumulation (bottom) overlapped with the area affected by siderosis. [Courtesy of Luka Kulic, Roche.]
This death was attributed to trontinemab. The drugs in its class are thought to largely sidestep CAA by crossing the BBB within capillaries, where TfR is most highly expressed, whereas CAA accumulates along larger vessels. So what gives? Brain shuttle developers discussing the findings at CTAD commented that while the lion’s share of trontinemab does cross via capillaries, some still manages to enter the brain via larger vessels, including arterioles and arteries. This could explain how trontinemab may have provoked a hemorrhage in this case, although the mechanism remains poorly understood.
“This shows that even if you deliver the antibody to the whole brain via TfR, you can still have consequences, especially if vascular health is poor,” said Costantino Iadecola of Weill Cornell Medical College in New York. He said that in this case, the superficial siderosis spotted on the screening MRI was likely “the tip of a very big iceberg,” and a slew of other vascular problems likely existed beneath the surface. This is especially likely given that the subsequent macrohemorrhage occurred in a different part of the brain, he said.
Going forward, Iadecola thinks scientists may need to go beyond excluding people with superficial siderosis, which is a potential indicator of severe CAA and other underlying cerebrovascular problems. Screening could include a comprehensive neurovascular assessment that takes into account several MRI measures, such as superficial siderosis, white-matter hyperintensities, microbleeds, and enlarged perivascular spaces. Perhaps an algorithm could incorporate all of these indices to calculate a neurovascular health score, he suggested.
At CTAD, Kulic proceeded by presenting data on other safety-related outcomes. TfR-targeted drugs come with a risk of anemia, due to high expression of TfR by reticulocytes. One way to deal with this this is to soften the antibody’s effector function, as Denali scientists have done with their antibody transport vehicle (ATV). For its part, trontinemab spares reticulocytes by way of steric hindrance, whereby engagement of the TfR-specific Fab temporarily blocks access to the antibody’s effector domain. In Madrid, Kulic reported that 13 percent of participants in Part 1 and 8 percent of participants in Part 2 have developed anemia, which was mild and went away. Roche decided to reduce the number of blood draws in Part 2 and is expecting fewer cases as a result, Kulic said.
What about ARIA? Kulic reported that, as was seen in the trial’s Part 1, few participants in Part 2 developed ARIA-E, despite receiving the two highest doses. So far, two people, both in the 1.8 mg dose group, had ARIA-E in this second, ongoing part of the study. Both cases were mild and asymptomatic, he said.
To Kulic’s mind, these ARIA rates are low given how robustly trontinemab lowers amyloid. At CTAD, he reported amyloid-PET data out to 28 weeks from Part 1 of the study (image below). Participants started with amyloid levels around 120 centiloids. By week 12, those in the 1.8 mg and 3.6 mg groups had lost 62 and 89 CL, respectively, bringing a majority of participants in the 3.6 mg group into the realm of amyloid negativity, below the 24 CL cutoff. By week 28, most participants in both these groups were below the cutoff, having lost an average of 84 and 107 CL, respectively.
Going, Going, Gone. In people on 1.8 mg (dark blue) or 3.6 mg (purple) of trontinemab, plaque load dropped by 12 weeks. By 28 weeks, most participants in both groups had crossed into amyloid negativity. Amyloid-PET scans of one person show removal (right). [Courtesy of Luka Kulic, Roche.]
Finally, Kulic reported preliminary CSF biomarker data from Part 1. At week 25, CSF total tau, p-tau181, and neurogranin had dropped dose-dependently, with concentrations of each falling by around 30 percent in the 3.6 mg dose group. This was not statistically significant owing to the small sample size.
Taking questions after his talk, Kulic said that because the 3.6 mg dose caused no more ARIA, anemia, or other adverse events than 1.8 mg, the study moved on to testing a higher dose. The 3.6 mg dose had not reached a plateau on the dose-response curve for amyloid removal, suggesting room for improvement, he said.
Iadecola agrees that trontinemab’s effect on amyloid-PET and CSF biomarkers looks promising. He cautions the studies were too small to draw conclusions about ARIA rates. One thing is clear: “Trontinemab is not going to be immune from causing ARIA,” he said.
Still, excitement about this data, and optimism about other TfR-based therapies coming down the pike, was palpable at CTAD. Other contenders are moving through preclinical studies, with trials in sight. Denali Therapeutics, based in South San Francisco, recently reported that its ATV delivered anti-Aβ antibodies throughout the parenchyma of 5xFAD mice. It features reduced effector function to protect reticulocytes, which the scientists believe suffices to rally microglia to take down Aβ (Pizzo et al., 2024).
Per-Ola Freskgård of BioArctic thinks compromising the ATV’s effector function might hobble its efficacy against Aβ. His company is developing what they call the BrainTransporter (BT), a construct that retains full effector function. BT includes a single chain variable antibody fragment that binds a novel site in TfR, Freskgård told Alzforum. The binding site is in the protease-like domain of TfR, which is positioned near the cell membrane.
In his talk accepting the CTAD Lifetime Achievement Award in AD Therapeutic Research, BioArctic’s Lars Lannfelt flashed one data slide showing how the BT dramatically improved an anti-Aβ antibody’s passage into an amyloid-ridden, 5xFAD mouse brain, and its engagement with plaques even in deep subcortical regions (image below). Lannfelt referred to the BT and similar TfR-based strategies as “the future” of Aβ immunotherapies.
BioArctic BT. Compared to an unmodified anti-Aβ antibody (green, left), a BT-equipped antibody better penetrated the brain of a mouse model of amyloidosis, where it engaged plaques in the brainstem and thalamus (bottom right). [Courtesy of Lars Lannfelt, BioArctic]
The week after CTAD, Freskgård presented preclinical findings with the BT at the Protein & Antibody Engineering Summit in Barcelona. There, he reported that the BT construct boosted passage across the BBB by 70-fold relative to an unmodified antibody, and did so without harming red blood cells. The BT construct rapidly and broadly penetrated the brain parenchyma, reaching deep subcortical structures, according to BioArctic. In April, the company signed a research evaluation agreement with Eisai to pursue development of “BAN2802,” an undisclosed AD drug equipped with BT technology. Freskgård would not disclose whether lecanemab or a different anti-Aβ antibody is being used in the BT construct.
ALIA-1758 is yet another BBB-crossing, investigational anti-Aβ immunotherapy. Developed by Aliada Therapeutics, it features an antibody trained against pyroglutamate Aβ equipped with an active transport system that was originally developed by Johnson & Johnson. It targets both TfR and CD98—receptors expressed by brain endothelial cells—for passage into the brain. ALIA-1758 is being tested for safety in healthy volunteers (see ClinicalTrials.gov). Abbvie recently shelled out $1.4 billion to acquire Aliada and carry ALIA-1758 into future trials.—Jessica Shugart
Donanemab: Small Tweak in Titration, Big Gain in Safety?
Seeking ways to improve the safety of anti-amyloid antibodies, John Sims of Eli Lilly & Company presented a simple option to do so for donanemab at this year’s Clinical Trials on Alzheimer’s Disease conference, held October 29-November 1 in Madrid. Starting at a low dose, and stepping it up over the first four months, cut the rate of ARIA-E by 40 percent, generating fewer severe cases to boot, Sims told the CTAD audience. The difference was biggest for APOE4 homozygotes, whose risk dropped by two-thirds, almost to the level in heterozygotes. Efficacy at clearing amyloid remained the same. Sims said Lilly will submit the data to global regulators to consider updating the label.
Gil Rabinovici of the University of California, San Francisco, called the data exciting. Also in Madrid, Rabinovici presented newly minted appropriate use recommendations for donanemab. They are similar to those for aducanumab and lecanemab, except that they call for more rigorous MRI criteria to exclude patients at higher risk for ARIA. A paper detailing the AUR is currently under review.
Rabinovici noted the manuscript might be updated to include Lilly’s modified titration protocol. Meanwhile, UCSF is already instituting the new titration scheme Sims presented at CTAD, he told Alzforum.
Move Over, Vial. In a new titration protocol (bottom), one vial of antibody moves from the first dose to the third; total donanemab exposure in the first six months remains identical to the standard protocol (top). [Courtesy of Eli Lilly.]
Simple Change, Big Payoff
In the Phase 3 Trailblazer-Alz2 trial, about a quarter of people on donanemab developed ARIA-E, with a fourth of those having symptoms such as headaches or confusion. The risk was highest in APOE4 homozygotes, with 40 percent of them experiencing edema (Jul 2023 conference news).
Lilly ran the Trailblazer-Alz6 trial to test different titration schemes in hopes of mitigating this. After all, the company is rolling out clinical use of donanemab at a time of intense scrutiny for anti-amyloid immunotherapy, and while their antibody is before regulators abroad.
Trailblazer-Alz6 enrolled 843 people, randomized to one of four dosing arms. The first tested the standard protocol, in which participants received three monthly doses of 700 mg, then 1,400 mg monthly thereafter. Arm 2 used a stepwise titration, with participants receiving doses of 350, 700, and 1,050 mg in the first three months (image above). In the third arm, participants had a first dose of 700 mg, skipped a month to let their brain acclimate, then went to 1,400. In the final arm, participants got 350 mg biweekly for the first three months, 700 mg biweekly in the fourth month, and 1,400 monthly thereafter. The goal there was to keep doses lower by spreading them out, with the theory that higher antibody concentration might provoke ARIA. In all four schemes, the cumulative donanemab exposure after four months was the same.
The scientists compared ARIA rates after six months, because in previous trials 90 percent of ARIA had occurred by then. As expected, the standard protocol produced the same amount of ARIA-E as in trials, 24 percent. Both skipping a dose, or spreading out doses, nudged down ARIA-E to about 18 percent, but this difference was not significant. The stepwise strategy, however, dropped the overall rate nearly in half, to 14 percent. For comparison, this is similar to the 12 percent ARIA-E rate reported for lecanemab.
Erasing Excess Risk. Under the standard titration protocol (blue), more than half of APOE4 homozygotes got ARIA-E; under a stepwise protocol (red), their risk resembled that of other genotypes. [Courtesy of Eli Lilly.]
For the 21 APOE4 homozygotes on the stepwise protocol, the change slashed their ARIA rate from 57 to 19 percent, near to the level in heterozygotes and noncarriers (image at right).
When ARIA did arise, it was less serious, Sims said. Three percent of people on stepwise titration had symptomatic ARIA-E, compared with 5 percent on the standard protocol. No ARIA-E cases were rated severe on MRI, compared with 2 percent severe on the standard protocol.
There were also fewer signs of the microhemorrhages known as ARIA-H. These often occur after edema; with the stepwise titration, the rate of concurrent ARIA-E and -H dropped from 16 to 10 percent. Rates of superficial siderosis, a sign of brain bleeds, halved, from 13 to 7 percent.
Despite this better safety overall, one person on the stepwise protocol died from ARIA. The person was an APOE4 carrier who had mild ARIA-E on the 24-week MRI scan, and went to the emergency room a week later with seizures and partial paralysis. There, the person was misdiagnosed as having a stroke and given the clot-buster tenecteplase, which led to a large brain bleed. Misdiagnosing ARIA-E as stroke, and giving tissue plasminogen-activator drugs, is emerging as a main cause of death for patients on amyloid immunotherapy. Most medical centers that prescribe anti-amyloid antibodies now have procedures in place to prevent this from happening. Alas, patients may show up at different ERs in case of crisis or when they travel. “Hospitals will have to learn not to accidentally kill Alzheimer’s patients on these antibodies by giving them thrombolytics,” Lawrence Honig of Columbia University, New York City, said with characteristic bluntness.
Meanwhile, the slower titration did not diminish plaque removal. The stepwise protocol cleared an average of 56 centiloids per person, versus 59 on the standard protocol, and it lowered plasma p-tau217 as much as did standard treatment. Cognition was not assessed in this trial, which is still ongoing, with participants being followed to 76 weeks, Sims noted.
The results suggest ARIA might be triggered by the initial contact between anti-amyloid antibodies and vascular plaque, Sims said. By keeping the first doses low, these reactions can be ameliorated, as the vasculature has slightly more time to adjust to incoming antibodies. “The first dose is important,” he said in Madrid.
AUR Emphasize Caution
To aid doctors who are now beginning to prescribe these antibodies, the clinician-researchers who had developed previous AURs for aducanumab and lecanemab have now issued recommendations for donanemab, as well (Aug 2021 conference news; Aug 2022 conference news; Apr 2023 conference news).
The group has expanded to 12 members. In addition to Rabinovici, they are: Paul Aisen of the University of Southern California in San Diego; Liana Apostolova of Indiana University School of Medicine in Indianapolis; Alireza Atri of Banner Sun Health Research Institute in Sun City, Arizona; Jeffrey Cummings of the University of Nevada, Las Vegas; Steven Greenberg of Massachusetts General Hospital, Boston; Suzanne Hendrix of Pentara Corporation, Salt Lake City; Ron Petersen of the Mayo Clinic in Rochester, Minnesota; Stephen Salloway of Butler Hospital in Providence, Rhode Island; Suzanne Schindler of Washington University, St. Louis; Dennis Selkoe of Brigham and Women’s Hospital, Boston; and Michael Weiner of the University of California, San Francisco.
Every anti-amyloid antibody is different, hence the donanemab AUR address two unique aspects of this drug: in its Phase 3 trial, scientists used baseline tau PET scans to determine eligibility, and they stopped dosing after a given person’s brain amyloid load had dropped below a preset threshold. For clinical practice, the AUR do not recommend tau PET. They do note that, if a scan is available, it can be used to estimate the benefit a person might experience from treatment. Regarding how long to treat, the AUR suggest clinicians consider stopping donanemab based on a visually negative amyloid PET scan taken after 12 to 18 months of infusions.
On safety, Rabinovici said the donanemab recommendations are meant primarily for physicians who have limited experience with this class of drugs. As such, the AUR are more stringent than the FDA label, erring on the side of caution. This is especially important during this sensitive initial period of learning how amyloid immunotherapy performs in the real world beyond clinical trial sites.
For example, the AUR suggest not treating patients who also take anticoagulants, because too few people in the Phase 3 trial were on these drugs to be able to assess donanemab’s risk in them. This recommendation will be subject to change as more data emerge, Rabinovici said. On the other hand, the AUR do not restrict anti-platelet drugs such as aspirin, since those were common in the trial and not associated with increased risk.
Both suggestions are in line with the lecanemab AUR. For lecanemab, there was evidence that anticoagulants amped up the risk of large brain bleeds and death in the Phase 3 trial (Jan 2023 news).
In another divergence from the label, the donanemab AUR recommend not treating patients who have even one area of superficial siderosis in their brains at baseline. These iron deposits, a relic of past bleeds, can indicate cerebral amyloid angiopathy, which amplifies the risk of ARIA. The Phase 3 trial had allowed one baseline area of superficial siderosis. However, an analysis by Greenberg linked this to a doubled risk of ARIA-E (Nov 2023 conference news). Others have found baseline siderosis in people who died on amyloid immunotherapy (Jan 2024 news; Jun 2024 news; Aug 2024 conference news). And also at CTAD in Madrid, trontinemab—the vanguard of a hopefully safer class of anti-amyloid antibodies to come—was shown to have been associated with a macrohemorrhage death likely linked to a baseline area of superficial siderosis, as well (see previous story in this series).
“We agreed unanimously that patients with baseline superficial siderosis should be excluded from treatment pending further safety data,” Rabinovici wrote to Alzforum. The lecanemab AUR have the same stricture. Moreover, Rabinovici noted that Lilly excludes people with baseline siderosis in its new trial of remternetug. “The field as a whole seems to be moving toward this position,” Rabinovici said.
In addition, if more than one area of superficial siderosis crops up during treatment, the AUR recommend stopping the drug. Some members of the AUR group thought even one occurrence was reason to stop, Rabinovici noted.
Scientists in Madrid held a spirited debate about how conservative to be on safety, and whether the FDA prescription insert or the AUR better guide clinicians. For example, both the donanemab and lecanemab AUR recommend against treating people with autoimmune disorders such as lupus, or with prior strokes. Honig has given lecanemab to patients with lupus, or who had recovered from a stroke years ago, and said they are doing well so far. He argued that because there is no clinical evidence for increased risk, the drug should not be withheld from these patients. “I believe in patient autonomy. Every patient should be offered a drug if it’s reasonable to do so, and the patient should consider the risk,” Honig said. In general, because the FDA label is more closely based on the clinical trial results than are the AUR, Honig prefers them.
Meanwhile, Salloway and others advocated for following the AUR. Salloway noted that he has heard from patients on autoimmune treatments who had severe, life-threatening ARIA. “Regulators and clinicians in some parts of the world are very concerned about safety,” he said.
What guidelines are memory clinics following? Previously, Alzforum found mixed results. Many clinics adhere strictly to the AUR. Others consider the label to be the firmer guide (Jan 2024 news). The issue is likely to stay in flux as more clinics begin using these drugs and real-world data comes in.—Madolyn Bowman Rogers
Leqembi: Side Effects No Worse in Clinical Use Than They Were in Trial
Lecanemab has been in clinical use in the U.S. for nearly two years, and in Japan for not quite a year. How is it going? At the Clinical Trials on Alzheimer’s Disease conference, held October 29 through November 1 in Madrid, speakers offered a snapshot. In the U.S., about 9,000 people have been treated, most of them white city dwellers. Clinicians are following the recommended schedule of MRIs for safety monitoring, and ARIA rates have been comparable to those in the trials, despite these real-world patients having more underlying health conditions than did trial participants.
In Japan, clinical use is climbing faster, with about 4,500 people now on the drug. ARIA rates in Japan are half those in the U.S., which was also the case in the Phase 3 Clarity trial.
Meanwhile, use in other countries is beginning, with lecanemab approved this year in China, South Korea, Hong Kong, Israel, the United Arab Emirates, and the United Kingdom. One problem in the UK: The country's National Health Service will not cover the cost, meaning access will be limited to wealthier people.
Not all countries are giving it the green light, however. In July, the European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) said no to the drug; in October, Australia's did. In August, the U.K. approved lecanemab but, striking a compromise, excluded APOE4 homozygotes, who have the highest risk of ARIA. Also last summer, drug maker Eisai asked the CHMP to reconsider, and today, on November 14, it did just that. Following the UK's example, EMA is now recommending that the European Commission approve lecanemab except for APOE4 homozygotes. At CTAD in Madrid, one talk suggested that non-carriers and heterozygotes reap a greater benefit from lecanemab, while having a lower risk (see below).
The other approved anti-amyloid antibody, donanemab, went on the market in July and has begun to enter clinic use, with around 700 people in the U.S. on it so far (see next story in this series).
Heading Up. The number of lecanemab infusions (colored dots) at Washington University, St. Louis, has grown steadily over a year. Most ARIA-A occurs around the fifth infusion. [Courtesy of Joy Snider, WashU.]
In the U.S., lecanemab has been on the market longest. In Madrid, Marwan Sabbagh of the Barrow Neurological Institute in Phoenix offered a bird’s eye view, by showing administrative claims data from the Komodo Health research database, which collects these records from more than 150 insurers representing 165 million patients. The data covered the period from January 2023, when lecanemab obtained accelerated FDA approval, through June 2024.
This analysis cohort comprised 3,155 people who had received at least one lecanemab infusion and had health insurance, i.e., about one-third of everyone treated in the U.S. In this group, 84 percent were non-Hispanic white, 93 percent lived in urban areas. Their average age was 75; 56 percent were women. Sixty percent were diagnosed as having mild cognitive impairment, indicating that the majority of the cohort were at an early disease stage, as expected. Four percent were taking anticoagulants, considered a risk factor for more serious side effects (Jan 2023 news; Aug 2023 conference news).
More than 90 percent of these patients started lecanemab after October 2023, when usage took off in the U.S. On average, it took nearly five months from their confirmatory amyloid test to starting infusions, indicating that the process remains slow. The average time on lecanemab in this early cut of real-world data was 129 days, or a little over four months. Most patients received their doses and scans per protocol, with the average time between infusions 16 days, and the first MRI done at 47 days, after the fourth infusion. “Lecanemab is being used appropriately,” Sabbagh concluded.
Sabbagh also noted that 85 percent of patients were still taking lecanemab at their four-month follow-up appointments. “Patients are highly motivated to stay on these drugs once they understand the consequences of treatment and nontreatment,” Sabbagh said. He has about 50 patients on lecanemab.
Reports from individual medical centers add detail to this broad-brush picture. Lawrence Honig of Columbia University, New York City, previously reported that among the first 122 people treated at Columbia, 7 percent developed ARIA-E, fewer than expected (Aug 2024 conference news). In Madrid, Honig updated the numbers.
By now, 162 patients have received an average of 13 lecanemab infusions each. Honig noted that many of them live far away, some even in Europe. They go to local infusion centers and MRI sites. As was seen in the Komodo Health database, 90 percent are non-Hispanic whites, and 60 percent have an MCI diagnosis. Their average age is 73. Most were genotyped for APOE, with 44 percent being E4 heterozygote and 14 percent homozygote. Typically, amyloid positivity was confirmed by cerebrospinal fluid biomarkers. Honig prefers that to PET, because a CSF analysis includes three biomarkers and gives him greater confidence in the diagnosis.
How have they fared? Eleven percent developed ARIA-E, about the same rate as in the Clarity trial. ARIA-E was typically mild and asymptomatic. In two cases, clinicians continued giving infusions, while in 14 others, dosing was paused until the edema cleared up. One person discontinued treatment due to ARIA-E, two due to ARIA-H. There have been no deaths since the previously reported case.
ARIA being quite manageable is notable, Honig said, because these patients have more co-morbidities than did trial participants. So far, Honig has treated one person on anticoagulants, three with prior cortical strokes or bleeds, and four with vascular malformations. He has also treated two patients with pacemakers, devices that can be affected by MRI scans. Radiologists are able to handle this, Honig said, often by having a cardiology nurse present.
His talk inspired a lively debate in Madrid over how conservative clinicians should be during this initial transition to immunotherapy when they make decisions about the treatment eligibility of the person across the table from them (Nov 2024 conference news).
Both Sabbagh and Honig emphasized that their institutions have put protocols in place to ensure that MRI monitoring is done appropriately, with infusions and scans prescribed in sets. For each set, the prescribing neurologist must sign off that the MRI has been done before a patient can receive their next infusion. “We instituted that as a solid stop. There has not been one violation,” Honig said.
Other health systems report similar experiences. Sutter Health in California employs more than 100 neurologists serving 3.5 million patients, and has thus far treated 234 people with lecanemab, according to neurologist Shawn Kile. All were screened either at one of the system’s two memory clinics, or by general neurologists using a review board. Neurologists followed the published AUR, with few exceptions, Kile said. All patients had APOE genotyping; 133 were heterozygote, 14 homozygote. Recipients’ average age was 70; 30 percent had mild dementia, the rest MCI.
So far, 15 patients have had ARIA-E; at 6 percent, this is half the rate of the clinical trial. Three of them had ARIA that was severe on MRI and caused confusion and headaches. One person had a lasting neurological deficit, a loss of vision in one-fourth of his or her visual field. Three people who stopped lecanemab due to ARIA developed a macrohemorrhage one to three months later, suggesting that side effects can occur with a delay. However, there were no deaths related to lecanemab treatment.
Likewise, Joy Snider of Washington University, St. Louis, offered an update from her medical center. At the beginning of the year, clinicians there had treated 68 people (Jan 2024 news). By a July 2024 data cut, the number of patients had risen to 186. As at other centers, nearly all were non-Hispanic white, with an average age of 74. Similar to in the trial, 13 percent developed ARIA-E, two-thirds of whom were mild and asymptomatic. Seven people had symptoms, two of those severe enough to require hospitalization. Notably, the majority of ARIA occurred around the fifth infusion; this was also seen at other institutions (image above).
Restricted Use in the U.K., Growing Numbers in Japan
With the U.K. limiting lecanemab use to non-homozygotes, Richard Perry of Imperial College London, working with Eisai scientists, showed how removing this group from the Clarity trial data would affect trial results. Without E4 homozygotes, there were 1,521 participants. Amyloid removal was the same as in the full cohort, at 59 centiloids per person. Cognitive outcomes were slightly better, with decline on the CDR-SB slowed by 33 percent, versus 27 percent in the full cohort, and other measures similarly nudged. The difference in ARIA was more notable. ARIA-E was lower by a fourth, from 13 to 9 percent, as was ARIA-H, falling from 17 to 13 percent. The rate of microhemorrhages nudged downward from 14 to 10 percent, superficial siderosis from 6 to 4 percent.
The EMA's CHMP committee cited Perry's analysis in its reconsideration, saying that, for this restricted group, the benefit of lecanamab treatment outweighed the risks (EMA announcement).
In Japan, lecanemab was approved in September 2023, and received insurance coverage at the end of that year. Thus, it has been in clinical usage for about 10 months. In Madrid, Takeshi Iwatsubo of the University of Tokyo told the audience that more than 600 clinics and 900 doctors now prescribe it. Iwatsubo noted that lecanemab use is restricted to major medical centers able to manage the multiple needs of patients, including MRI monitoring.
Another wrinkle: APOE genotyping is required for lecanemab use, but is not covered by insurance. As a workaround, patients enroll in a registry maintained by academic centers, which takes care of genotyping.
An estimated 4,500 prescriptions have been filled in 2024 so far, Iwatsubo said. About 80 percent of patients have mild cognitive impairment, the rest have mild dementia. Most are in their 70s, and almost two-thirds are women. Confirmation of amyloid positivity is done by PET two-thirds of the time, cerebrospinal fluid the rest.
Curiously, in the Clarity trial, ARIA occurred at only half the rate in Asian participants compared with other geographic regions in this global study. There were only 10 cases among 153 participants, for a rate of 6.5 percent. For the 88 Japanese participants, the rate was even lower, at 4.5 percent. In clinical usage so far, ARIA rates have matched that, with around 5 percent of patients developing ARIA-E or ARIA-H, Iwatsubo reported.
Though the rollout has been smooth so far, Iwatsubo noted hurdles to future expansion. In this country of 124,000 million people, with its aging society, only about 2,400 physicians are licensed to prescribe anti-amyloid antibodies. There is also a shortage of infusion sites. Among the 10 million Japanese people with MCI or AD, about 1 million are estimated to be eligible for treatment. Only a small proportion of them will be able to get lecanemab, Iwatsubo said, adding that more treatment options for AD are needed.—Madolyn Bowman Rogers
Finally, Therapeutic Antibodies Start to Reduce Tangles
The fifth time may be the charm for antibodies targeting tau. At the 16th Clinical Trials on Alzheimer’s Disease conference, held October 29 to November 1 in Madrid, Brussels-based UCB Pharma presented the first signal that a monoclonal antibody might be slowing the progression of tau pathology in the brain, at least in some people. Its drug, bepranemab, did not improve symptoms in the full trial population, but it did offer a signal of clinical benefit in people with low baseline tau levels and no ApoE4. On balance, researchers at CTAD were hopeful that bepranemab, or similar candidates, could mark the beginning of a new Alzheimer's treatment strategy.
“It’s an important milestone for the field,” Adam Boxer, University of California San Francisco, told Alzforum. Not only are tau antibodies promising as treatments, Boxer said, but the results also suggest that tau seeding and propagation through the brain plays an important role in driving Alzheimer's clinical symptoms. This would open the door to better understanding the disease mechanism. “There is now clear experimental evidence–not just in animals or cell culture—that tau transmission extracellularly is doing something,” Boxer said.
“I think it’s really promising finally to see an antibody against tau moving the needle,” Rakez Kayed, University of Texas Medical Branch, Galveston, told Alzforum.
Immunotherapy against tau, given either alone or in combination with anti-amyloid antibodies, has been a goal of many companies for years. That said, four earlier antibodies showed no effect on either tau clearance or symptoms in clinical trials. They all targeted the protein’s N-terminal region, which may have been the reason they failed, said bepranemab developer Martin Citron of UCB, Brussels. While they engaged their targets well, later data suggested that this end of tau is cleaved off before tau aggregates, and likely irrelevant to the disease process.
Most of the current generation of investigational tau antibodies, including bepranemab, bind at or near a central part of the protein known as the microtubule binding region. The MTBR is important for tau aggregation, and some scientists think it may be the source material of tau seeding in the brain (Mar 2021 conference news; Apr 2018 conference news).
Most discontinued anti-tau antibodies (red) targeted tau’s N-terminus. Of the antibodies currently in trials (green), bepranemab and E2814 presented results at CTAD, while JJ-63733657/posdinemab and PRX005/BMS0986446 presented trial designs. Active vaccines in blue. [Courtesy of Translational Neurodegeneration.]
In their October 31 talks at CTAD, Citron and Matthew Barton, UCB, presented the first data from the company’s Phase 2 TOGETHER trial in 466 people with prodromal to mild Alzheimer's disease. The trial missed its primary endpoint of improving cognition and function as measured by the CDR-sum of boxes in the full trial population.
At the same time, bepranemab slowed tau accumulation measured by PET by up to 58 percent compared to placebo after 80 weeks of treatment. Moreover, the researchers found an encouraging hint when they split trial participants into two types of subgroups: low versus high baseline tangle load and ApoE4 mutation carrier versus noncarrier. When they removed ApoE4 carriers with high tau from the equation—presumably the most advanced cases in this trial population—the scientists found that bepranemab appeared to have halved cognitive decline on the ADAS-Cog, a secondary outcome, compared with placebo. Alas, the subgroup with high tau at baseline appeared to do worse on treatment than placebo.
Randall Bateman, Washington University, St. Louis, welcomed the results in the low-tau group. He noted that anti-amyloid antibodies also tend to work best at this stage of disease. “It may be now a question of can you find the right population to treat with this kind of antibody, and get the right timing so that you can prevent the growth and spread of tau pathology?” he told Alzforum.
Citron told Alzforum that UCB is still deciding whether it will evaluate bepranemab in another Phase 2 trial specifically in an early tau population before moving to a pivotal Phase 3 study.
Michelle Farrell, Massachusetts General Hospital, Boston, said that the discrepancy in the bepranemab trial between the high and low tau groups support Keith Johnson’s hypothesis of a “cataustrophe,” whereby tangles accumulate in the medial temporal lobe before symptom onset, then, somehow in conjunction with amyloid plaques, suddenly spread more quickly across the cortex and trigger a cascade of subsequent pathologies (Apr 2022 news; Johnson et al., 2020).
Treating people before this happens might be key, Farrell told Alzforum. “There may be an element where once things get moving, it’s hard to stop.” She added that low tau pathology burden does not always correspond with mild symptoms, hence PET or fluid biomarkers might be more useful than clinical measures in determining whether a person is likely to benefit from immunotherapy. “That is an important component when we’re thinking about who we should be targeting for these trials,” Farrell said.
While bepranemab’s results in the low tau subgroup made sense to scientists at CTAD, the trial’s finding that the drug had no measurable effect in people with the ApoE4 genotype mystified them. Scientists still debate whether ApoE4 affects tau propagation or tau’s role in driving clinical symptoms. Mouse and some human experimental data suggest that the isoform accelerates tau seeding and tau-mediated neurodegeneration independent of Aβ pathology. (e.g., Shi et al. 2017; Koutsodendris et al., 2023). ApoE4 carriers also start depositing amyloid plaques up to a decade earlier than noncarriers, hence their AD pathogenesis might have been more advanced. Barton did not relate participants’ outcomes to their baseline amyloid load, but Citron said these analyses would be done.
Other drug developers are trying to get a handle on the proposed cataustrophe as well. Monitoring tau spread is a distinctive ambition of the ongoing Autonomy Phase 2 trial for posdinemab. This antibody targets phosphorylated tau near the p217 site in tau’s proline-rich mid-domain region just upstream of the MTBR. At CTAD, David Henley, Janssen, Titusville, New Jersey, reported that this trial, which began in 2021, has enrolled 523 people with intermediate levels of tau pathology in their temporal lobes. The trial randomized participants into high dose, low dose, and placebo groups and will treat each person once a month for two to four years. Using tau PET, the study is tracking how each participant’s tau spreads into regions of their brain where it previously had not been seen, rather than only measuring the overall tau burden in a participant’s brain. The hope is that tangles will spread less in people receiving antibody than placebo. The trial will also assess clinical measures.
Janssen screened participants using a Ptau217 blood test (see upcoming story in CTAD series), which Henley said halved the number of required PET scans.
Companies besides UCB and Janssen are also going after tau. Kristin Wildsmith, Eisai, Nutley, New Jersey, presented results for an open-label, ascending-dose trial of E2814, an antibody that has two separate binding sites in the MTBR. Completed in May 2024, this study was conducted in eight people with autosomal-dominant Alzheimer's who already had mild to moderate symptoms at the study’s start.
In her CTAD presentation, Wildsmith added further data to an earlier presentation by Jin Zhou of Eisai on this trial, which was designed to assess safety and target engagement (Aug 2023 conference news) The participants received the drug for up to 26 months, though five of them dropped out along the way. Comparing their course to the disease progression seen in otherwise matched participants in the DIAN observational study (DIAN-OBS), Wildsmith estimated that E2814 cut the CSF pTau217 concentration in half over the course of two years of treatment. Three people remained in the trial long enough to undergo brain scans, which trended toward a reduction in tau PET, though Wildsmith noted that some of this may be due to atrophy.
Bateman, who leads the DIAN initiative, acknowledged that the numbers are too small to be conclusive, and an Eisai spokesperson confirmed that the data largely serve to inform current E2814 trials. The Phase 2/3 NexGen trial run by the Dominantly Inherited Alzheimer’s Network Trials Unit (DIAN-TU) has finished enrolling its 197 participants; results are expected in 2028. Another Phase 2 study in 90 participants with sporadic MCI due to AD started up in September 2024 and is currently enrolling.
Last but not least, CTAD saw mention of another Phase 2 trial of a tau antibody. Similar to E2814, BMS-986446 binds to tau’s MTBR, regardless of whether the protein is phosphorylated. The 18-month TargetTau-1 started enrolling people with mild cognitive impairment or mild Alzheimer's pathology in March 2024, Chris Van Dyck, Yale University, told the audience.
Thus far, all of the tau antibodies seem to be safer than anti-amyloid antibodies, which can cause ARIA. Bepranemab is an IgG4 that doesn’t fully activate the immune system; posdinemab, E2814 and BMS-986446 are IgG1s that have full effector function, i.e. activate microglia in hopes of more protein clearing.
Citron, and many others, believe that it is time to test tau antibodies in combination with amyloid antibodies. “We've been hammering away at amyloid for 30 years, and one thing we understand pretty well now is that, with amyloid clearance, we are maxing out slowing of disease progression at about 30 percent,” he said. At CTAD, a panel discussion featured debate about the promise and design challenges of combination trials. Notably, DIAN-TU’s NextGen trial and the new E2814 trial are already testing this tau antibody concurrently with lecanemab.
For his part, Boxer has been planning for some years to launch the Alzheimer’s Clinical Trial Consortium tau platform trial, aka ATP, to test two tau therapies alone or in combination with amyloid drugs (Aisen et al., 2021; Nov 2021 conference news). He hopes ATP will start up in 2025. In Madrid, Boxer said that ATP will initially enroll 750 people with late preclinical to prodromal Alzheimer's, with a primary endpoint of slowing tau accumulation as per PET over two years. The researchers will also look at blood and CSF biomarkers and clinical decline, and will add another 150 participants when they begin testing a second tau therapy. Boxer declined to say which drugs will be tested.
Kayez said that for all tau antibody development, researchers may want to look at postmortem brains to determine whether the antibody at hand is targeting all forms of aggregated tau or only those that can be seen on PET imaging. “Despite all the positives, I think important questions still need to be addressed,” he said.—Sara Reardon
Sara Reardon is a freelance writer in Bozeman, Montana.
Tau Modification Drugs Take a Hit with Negative Trial
While anti-tau antibodies are beginning to look promising (see previous story), small molecules that modify tau proteins haven’t yet fared well in clinical trials. At the 16th Clinical Trials on Alzheimer’s Disease conference, held October 29 to November 3 in Madrid, Eli Lilly’s negative trial was a setback for ceperognastat, the company’s inhibitor of O-linked-β-N-acetylglucosaminidase (O-GlcNAcase, aka OGA).
O-GlcNAcase is an enzyme that removes sugars from serine or threonine residues on thousands of nuclear, mitochondrial, and cytoplasmic proteins, including tau (see Zachara et al. chapter in Essentials of Glycobiology, 2022). These moieties seem to compete with phosphorylation at certain sites in tau, stabilizing the protein and making it less prone to form neurofibrillary tangles. In mouse studies, inhibiting OGA with small molecules has led to less hyperphosphorylation and subsequent NFT formation (Mar 2012 news; Mar 2024 news).
Sugar On, Sugar Off. Many mammalian proteins are modified by monosaccharides of O-linked β-N-acetylglucosamine. O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) add and remove O-GlcNAc, respectively. On some proteins, O-GlcNAc competes with phosphorylation for serine/threonine residues, and some proteins cycle between phosphorylated and glycosylated states. [Courtesy of Essentials of Glycobiology, 2022.]
For its Phase 2 PROSPECT-ALZ study, Lilly tested ceperognastat in 327 people with early symptomatic Alzheimer's, stratifying them into low/medium and high baseline tau groups. They treated both groups with either 0.75 or 3 mg doses or a placebo for 76 weeks.
Taking Out OGA. The rationale behind small-molecule OGA inhibition is that these drugs will decrease tau hyperphosphorylation and aggregation into neurofibrillary tangles (right). [Courtesy of Eli Lilly & Co.]
In his October 30 CTAD presentation, Lilly’s Adam Fleisher showed that ceperognastat missed its secondary outcome of reducing clinical symptoms in either group. On the contrary, the 3 mg group declined significantly faster on almost all cognitive measures compared with placebo. Both dose groups also had more serious adverse events, including cardiac and nervous system disorders and neoplasms. Treatment-related effects such as headaches and cardiac abnormalities were twice as likely to be severe in the high compared to the low-dose group.
Intriguingly, though, the primary outcomes told a different story. The treatment groups lost up to 45 percent less hippocampal volume over the 76-week period than did people on placebo. Moreover, the 3 mg group showed statistically significant slowing of tau accumulation in the temporal lobe, as per flortaucipir PET. Ceperognastat also improved plasma biomarkers, lowering phosphorylated tau217 levels and inflammation signals in the low/medium tau population.
In a statement to Alzforum, Lilly’s corporate communications said the company is continuing to monitor data from an extended safety study that will wrap up in February 2025. “Although the primary endpoint was not met, this study provided learnings with regards to the possibility of discordant outcomes of clinical and biomarker endpoints,” the company wrote. “Future studies, while currently not being planned, might be potentially considered.” Lilly added that it will “continue to explore potential treatments targeting [tau] pathology.”
Adam Boxer, University of California, San Francisco, told Alzforum the results fit with his concerns about small molecules targeting post-translational tau modifications. OGA drugs, he said, “have been around for a long time and so it’s interesting to see definitive results.” While the PROSPECT-ALZ results support the hypothesis that increasing glycosylation reduces tangle formation, the finding that patients did worse bodes ill for its clinical potential, at least as pursued currently.
Likewise, Rakez Kayed, University of Texas Medical Branch, Galveston, was unsurprised the trial result was negative. To his mind, targeting an enzyme that affects thousands of proteins throughout the body is a “nonstarter” due to potential off-target effects. “Even if it worked and met its secondary endpoints, the side effects could be massive,” he said.
OGA’s opposite, the O-GlcNAc transferase enzyme that attaches GlcNAc to protein serine and threonine residues, was spotted in the postsynaptic density of spines of excitatory neurons, and implicated in synaptic regulation (Lagerlöf et al., 2017).
Boxer added that it remains unclear whether the trial’s adverse events and worse clinical outcomes were due to off-target effects. “These are pretty dirty drugs, and you can't interpret the tau PET without the context of everything else going on,” he said. “Such a large attenuation of atrophy may mean it’s affecting many things in the brain.”
Whether OGA inhibitors would affect other tauopathies differently is still an open question. The Spanish company Ferrer is conducting a Phase 1 trial of a different candidate for progressive supranuclear palsy. While the mechanisms of tau accumulation in PSP likely differ from those in Alzheimer's, Boxer said, the drug might cause similar off-target effects in both diseases.
Ceperognastat is one of three OGA inhibitors in trials for Alzheimer's and the first to have reached Phase 2. Biogen’s BIIB113 wrapped up its Phase 1 trial last year, but the company didn’t answer a question about its future plans for the molecule (Mar 2024 news). A spokesperson told Alzforum that the company is “working to analyze both internal and external insights of the study.”
Asceneuron is developing an OGA inhibitor called ASN51. It began Phase 2 last month, with a safety and biomarker trial that plans to enroll 78 people and run through 2026.—Sara Reardon
Sara Reardon is a freelance writer in Bozeman, Montana.
Fully Loaded: Secondary Prevention Studies of Lecanemab, Donanemab
Anti-amyloid antibodies lecanemab and donanemab are in clinical use now, but that does not mean research on them has stopped. At the Clinical Trials on Alzheimer’s Disease conference, held October 29-November 1 in Madrid, speakers updated the audience on new or ongoing studies. For lecanemab, they focused on the secondary prevention trial AHEAD 3-45, now fully enrolled. They showed how plasma prescreening panned out, along with preliminary baseline data for this population. Likewise, donanemab’s secondary prevention trial Trailblazer-Alz3 finished enrolling; its baseline data showed that it captured a slightly different population than did AHEAD, with less genetic risk.
On new studies, Lilly scientists presented efforts to gather real-world data on donanemab’s effectiveness, and debuted the Phase 3 trial design for donanemab’s successor, remternetug, which patients can inject under the skin.
During CTAD, news broke that Eisai had completed its Food and Drug Administration submission for subcutaneous lecanemab. This formulation would be given as a weekly maintenance dose of 360 mg, after people have finished their intravenous course. Previously, Eisai had requested approval for IV maintenance dosing, as well (May 2024 news). And days after CTAD, news broke that European regulators reversed their July 2024 rejection of lecanemab, and are now recommending approval (EMA release).
Preclinical Populations. On the plaque accumulation curve (red), the A3 cohort (orange circle) is at an earlier stage than the A45 cohort (red circle). [Courtesy of Paul Aisen.]
Lecanemab Secondary Prevention Study Now Fully Enrolled
Lecanemab was approved for people with mild cognitive impairment or mild dementia due to Alzheimer’s, but trial data show it works better if given earlier. The AHEAD 3-45 study, a pair of sister trials, homes in on this point, testing lecanemab’s ability to delay the onset of symptoms. Both trials enroll cognitively healthy people with plaque in their brains; in A3, they have between 20-40 centiloids, in A45, they have more than 40 (Nov 2020 conference news).
Because finding people with preclinical AD requires testing huge numbers of healthy ones, trial leaders prescreened with a blood test, using the ratio of p-tau217 to non-phosphorylated tau217 to predict amyloid positivity. Previously, they had reported that this screened out two-thirds of potential participants (Aug 2021 conference news; Nov 2021 conference news).
In Madrid, Reisa Sperling of Brigham and Women’s Hospital, Boston, shared a final look at the screening process for AHEAD, which finished enrolling in October. The plasma p-tau217 ratio, when combined with the plasma Aβ42/40 ratio, age, and APOE genotype, identified people with amyloid plaque with an AUC of 0.95. Among the one-third of screened participants who were positive on this marker and went on to get an amyloid PET scan, around 60 percent qualified for the trial—a big improvement over the 25 percent success rate of PET scans before plasma prescreening, Sperling said.
Between the amyloid loads of 5 to 100 centiloids, the plasma p-tau217 ratio (p-tau217r) rose in tandem with plaque, enabling Sperling’s team to adjust the p-tau217r cutoff to select for different amounts of amyloid burden. When trialists used a higher cutoff to identify people specifically for the A45 study, 75 percent of those who passed plasma screening also qualified by PET. Most had plaque loads between 40 and 100 centiloids, but a few of these cognitively healthy adults had more, going as high as 160 centiloids, Sperling said.
At the other end of the scale, people who were amyloid-positive below 20 centiloids are being followed in the APEX observational study. Altogether, study personnel screened 20,721 people and performed 4,443 PET scans to find 1,621 participants; 448 are enrolled in A3, 1,173 in A45. By comparison, in the A4 study, 4,600 scans were needed to find 1,169 participants, a 25 percent success rate (Mar 2023 news).
Other scientists in Madrid likewise reported success with plasma p-tau217 prescreening—for example, in a trial of the anti-tau antibody posdinemab, this step halved the number of PET scans needed (Nov 2024 conference news).
Paul Aisen, University of Southern California, San Diego, presented baseline characteristics of the A3 and A45 trial populations. In A3, the average age is 68; in A45, 70. In both studies, almost two-thirds of participants are women; most are college-educated. Eighty percent have a family history of dementia, 73 percent carry at least one APOE4 allele. All have a baseline global CDR of 0. That said, around 10 percent of both cohorts have slight decline on the CDR-SB, with a score of 0.5 or 1 on this more sensitive measure, indicating the total AHEAD population is not completely asymptomatic.
Racial and ethnic minorities comprised more than a quarter of the screening population; alas, they were more likely to have negative amyloid scans, hence 90 percent of the final participants were once again white.
What about tau PET? Many participants had positive scans, Sperling reported. In the A3 group, tangle accumulation in the medial temporal lobe was common, less so in the neocortex. The A45 group had a higher tangle load in both the MTL and neocortex. Higher tangle loads correlated with subtle cognitive deficits on the PACC5 cognitive test, though the relationship was weak, at r=0.13 for MTL tau and 0.16 for neocortical tau, Sperling said.
The trials will run for four years. Aisen noted that in both, participants will be randomized evenly to placebo or lecanemab, and will titrate up, receiving 5 mg/kg for two months, 10 mg/kg thereafter. The main difference between the studies is that A3 participants, who have less plaque, will get a single monthly dose throughout the trial, while A45 participants will get doses every two weeks for the first two years, monthly thereafter. After four years, participants will transition to a 12-week, open-label extension, in which everyone will receive biweekly doses again, so that former placebo patients get effective plaque clearance, Aisen told Alzforum.
Because lecanemab is an approved therapy for early symptomatic AD, participants who develop cognitive problems during the trial may enter the OLE early. To qualify for that, participants must score higher than 0 on the CDR at two successive visits, and be judged to have MCI or mild dementia by a site clinician.
For A3, which is a Phase 2 trial, the primary outcome will be amyloid PET, with tau PET secondary and the PACC5 and other cognitive tests exploratory. For A45, a Phase 3 trial, PACC5 is the primary outcome, with amyloid and tau PET secondary. This trial also measures plasma and cerebrospinal fluid markers, additional cognitive tests, and participant self-reports.
Reaching Farther. The decentralized design of Trailblazer-Alz3 allowed researchers to recruit people from more U.S. states (right) than did the A4 trial (left). [Courtesy of Eli Lilly.]
Donanemab Secondary Prevention Study Tries Decentralized Design
Donanemab has its own prevention study, Trailblazer-Alz3, run by Eli Lilly. It finished enrolling in August 2024. Previously, the company had reported that participants will receive nine monthly infusions of donanemab or placebo, with the primary outcome being time to clinical worsening on the CDR. The trial will follow participants until 350 of them have progressed in this way (Aug 2024 conference news).
In Madrid, Lilly’s Karen Holdridge fleshed out details. To find enough participants, including people in remote areas, the trial used a decentralized design whereby initial cognitive screening happened via the modified telephone interview for cognitive status (TICS-m), and amyloid positivity was determined by a p-tau217 blood test. Holdridge noted that Lilly went out into community settings to find people, manning mobile research units and drawing blood at health fairs. Participants received tablets on which to take cognitive tests at home every six months. This design allowed Lilly to enroll people from 40 U.S. states. Compared with previous trials, that is better geographic representation, particularly in the American south and west (image above). “We cast a broad net,” Holdridge noted.
Screen failures were higher than in AHEAD 3-45, with about 80 percent of prospective participants disqualified by the plasma p-tau217 screen. In total, study personnel screened 63,124 people in the U.S. and Japan to find 2,196 participants. Curiously, 499 of them dropped out before beginning infusions, leaving 1,697 people in the trial.
Participants are 70 years old on average; two-thirds are women, and most have completed college. As in AHEAD 3-45, racial and ethnic minorities were more likely to be amyloid-negative, generating a final cohort of 90 percent non-Hispanic whites. Half the participants have an AD family history, lower than the 80 percent in AHEAD 3-45. All were genotyped for APOE; nearly half are APOE4 heterozygote, 8 percent homozygote, again lower than AHEAD’s numbers. Participants can choose whether to find out their genotype from a genetic counselor.
Earlier Population. In the Trailblazer-Alz3 cohort (left), participants with a CDR of 0.5 have less plaque (top) and fewer tangles (bottom) compared with CDR 0.5 participants in the Phase 3 Trailblazer-Alz2 trial (right). [Courtesy of Eli Lilly.]
Unlike in AHEAD 3-45, not all Trailblazer-Alz3 participants are cognitively normal at CDR 0. Because the TICS-m was used for screening, about one-third of the cohort is CDR 0.5, which is considered mild cognitive impairment. Even so, Holdridge said the methodology captured an earlier AD population than did donanemab’s Phase 3 trial, Trailblazer-Alz2.
This was borne out by baseline amyloid and tau PET scans done in substudies of 432 people opting for amyloid PET, 331 for tau PET. On amyloid scans, people who were CDR 0.5 had an average of 71 centiloids, similar to the 63 centiloids in the CDR 0 group. This compares with 103 centiloids for the CDR 0.5 group in Trailblazer-Alz2. On tau scans, those at CDR 0.5 averaged 1.1 SUVR—a little higher than the 1.04 in the CDR 0 group but lower than the 1.31 in the CDR 0.5 group in Trailblazer-Alz2 (image above). Scientists in Madrid agreed with Holdridge that this is a different population than other antibody studies have enrolled, with fewer genetic risk factors.
In addition to this prevention study, Lilly is running two trials to collect real-world evidence on donanemab’s safety and effectiveness. In Madrid, Lilly’s Ann Hartry described them. One, dubbed Platform for Early Alzheimer’s in Real Life (PEARL), invites clinicians in the U.S. to refer patients on immunotherapy to the study; screening and data collection are done remotely. Much of the data collection is passive, leveraging Medicare claims to gather demographics, diagnoses, test results, and treatments. The study will gather five years of retrospective data and five years of prospective data for each participant. Hartry categorized this as “low-touch” yet able to collect regulatory-grade data.
The other one, Trailblazer-Real US, will compare how patients on donanemab or standard Alzheimer’s care fare after five years. The primary outcome will be time to the first loss of independence. The study will also measure neuropsychiatric status and caregiver burden.
Donanemab was approved for clinical use in the U.S. in July, and is ramping up slowly, with an estimated 700 patients now on it. Joy Snider at Washington University in St. Louis noted that, as with lecanemab, institutions have to build infrastructure to start administering. These include hospital formulary approval, pharmacy and infusion center training, and electronic medical record order sets. At WashU, this will be finalized as of November 20, after which infusions can begin. Snider believes that, once available, some patients will opt for donanemab over lecanemab. “Many patients are looking for less frequent infusions, so the monthly [dosing] versus biweekly for lecanemab will be attractive, as is the idea that you can stop infusions after 18 months,” she wrote to Alzforum.
Remternetug Enters Phase 3
Donanemab may soon face competition from within Lilly itself. The company’s successor molecule, remternetug, targets the same amyloid species as donanemab but removes plaque faster while triggering fewer anti-drug antibodies or infusion reactions. Unlike donanemab, remternetug has been designed to be injected under the skin (Apr 2023 conference news).
In Madrid, Kevin Biglan at Lilly described a new, global Phase 3 trial, Trailrunner-Alz3. Enrollment began in October, with a goal of recruiting 1,200 people between 55 and 80 who have preclinical AD or mild cognitive impairment. To capture an early population, researchers have set cutoffs of 27 or higher on the MMSE, and below 6 on the FAQ. Screening for amyloid positivity will be by way of Lilly’s plasma p-tau217 test.
Like the donanemab prevention study, Trailrunner-Alz3 will be decentralized, where participants self-administer doses at home and will have options for where to get MRI scans. Cognitive tests will be sent to a single center to be rated, reducing variability.
Participants will take remternetug or placebo for 18 months, followed by an observation period during which participants will stay blinded and get cognitive assessments every six months. The primary outcome will be time to clinical worsening on the CDR, with other cognitive tests as secondary outcomes. The trial will include an amyloid and tau PET substudy with 400 participants. Once enough people have worsened, Lilly will lock the data and invite participants into an OLE, where the placebo group will get access to remternetug.
One detail of this study caused some consternation in Madrid. Biglan noted that the trial will not exclude people who have had previous amyloid immunotherapy, so long as that therapy concluded more than five antibody half-lives before screening. One audience member pointed out that because the trial does not measure baseline amyloid load, it might be hard to quantify remternetug’s effect in a mixed population where some participants have little plaque. Biglan said Lilly plans to “front-load” its PET substudy to get a better idea of baseline loads.
Remternetug is being tested in the Dominantly Inherited Alzheimer Disease Trials Unit’s primary prevention trial (May 2024 news; Aug 2024 conference news).—Madolyn Bowman Rogers.
Biomarkers Suggest Black and Hispanic People Less Likely to Have Amyloid
Despite higher rates of clinically diagnosed dementia among people from non-white racial and ethnic groups, historically they have been underrepresented in AD clinical trials. Efforts are underway to boost recruitment among these populations into clinical studies. They are successful inasmuch as black and Hispanic people are showing up for trial screenings. However, as scientists reported at the CTAD conference held October 29 to November 1 in Madrid, current trials still end up with too few participants from underrepresented groups because scientists are finding that their prevalence of amyloid positivity is significantly lower than that of whites, rendering many eligible for trial enrollment.
New screening data reported from ongoing and past trials indicated that, across racial and ethnic groups, blood biomarkers were faithful predictors of amyloid plaques lurking in the brain. However, in both cognitively normal and impaired populations, non-Hispanic white volunteers consistently had higher amyloid prevalence, resulting in their overrepresentation in amyloid-targeted trials. AHEAD 3-45, TRAILBLAZER-ALZ2 and 3, and New IDEAS were the main studies presenting this finding at CTAD.
Other studies suggest that people from underrepresented groups tended to have more comorbidities, and to live with a higher burden of social and economic strain, both of which may contribute to health disparities. How this factors into amyloid prevalence remains unclear, noted Michael Weiner of the University of California, San Francisco. Because studies are prone to selection bias, he said, it is difficult to know if differences in amyloid prevalence reflect those found in broader ethnoracial populations. Others believe that the lower prevalence of amyloid in black and Hispanic communities implies that other underlying pathological processes—such as vascular dysfunction—drive higher dementia rates in them, and they called for more research on those processes. Scientists at CTAD sounded highly motivated to figure out which physiological factors underlie differences in the way dementia develops across populations (see also Nov 2023 conference news).
In Madrid, Doris Molina-Henry of the University of Southern California in San Diego presented biomarker screening data from AHEAD 3-45, a pair of AD prevention studies that are evaluating lecanemab in cognitively normal people with intermediate (A3) or elevated (A45) levels of amyloid accumulation at baseline (Nov 2024 conference news). The trial finished randomizing its 1,621 participants in October. Finding them was a tall order, requiring the screening of 20,721 people. Twenty-seven percent of those screened came from underrepresented ethnoracial groups, approaching the 2023 U.S. Census Bureau estimate of 33 percent of the U.S. population being black or Hispanic. This was progress. For comparison, the previous A4 prevention trial, which also attempted to diversify recruitment, netted 14 percent non-Hispanic white.
Alas, among those who were subsequently enrolled in A3 and A45, the percentage of people from these underrepresented groups was only 15 and 11 percent, respectively. In Madrid, Molina-Henry attributed most of these screen failures to volunteers having negative AD blood biomarker tests.
The trial aimed to enroll cognitively normal people with 20 to 40 centiloids of amyloid based on PET scans for A3, or greater than 40 centiloids for A45. The screening process took nearly four years. Initially, it started with blood collection and cognitive testing at the first visit, followed by an amyloid-PET scan at visit 2. Once AD blood biomarkers became validated, the researchers introduced a plasma prescreening step—first with Aβ42/40 and later adding in p-tau217—hoping to reduce the number of negative PET scans. In the final months of screening, to complete enrollment for A45, the researchers raised the plasma cut-off to let in more people with elevated amyloid (image below).
Stages of Screening. The early months of screening for AHEAD3-45 used cognition and amyloid-PET. Adding plasma screening—first with Aβ42/40, then also p-tau217—drove down the number of people who were subsequently shut out due to a negative amyloid PET scan. [Courtesy of Reisa Sperling, Brigham and Women’s Hospital.]
The scientists had previously reported outcomes of plasma prescreening based on the Aβ42/40 ratio, adjusted for age and ApoE4 status (Molina-Henry et al., 2024). During this phase of screening, they set the algorithm to select a plasma biomarker threshold predictive of at least 11 centiloids on amyloid-PET—an intentionally low bar to maximize recruitment for A3, Molina-Henry told Alzforum. That revealed that people self-identifying as non-Hispanic white were more likely to cross this threshold for amyloid positivity than people from any of the other ethnic or racial groups. Among those whose plasma biomarkers made them eligible to move forward to a PET scan, about half subsequently tested positive via amyloid-PET. This was true across all ethnic and racial groups, suggesting that the relationship between plasma Aβ42/40 and amyloid-PET positivity held across groups.
Who Gets In? Non-Hispanic whites (purple line) were less likely to be ineligible based on plasma biomarkers than were people from other ethnoracial groups. [Courtesy of Doris Molina-Henry, University of Southern California, San Diego.]
At CTAD, Molina-Henry showed results of the next phases of screening, which incorporated plasma p-tau217 along with Aβ42/40. For this, the scientists set the algorithm to select participants with blood biomarker levels predictive of at least 18 centiloid of amyloid. Adding this additional biomarker weeded out more people, further reducing the number of unnecessary PET scans. But again, non-Hispanic white people were more likely to have this much amyloid than any other group. While 27 percent of non-Hispanic whites were "plasma-eligible" to move forward with amyloid-PET, only 19 percent of Hispanic white, 11 percent of Hispanic black, 19 percent of non-Hispanic black, and 15 percent of Asian people were. Among those who moved forward with a scan, there were no differences across ethnoracial groups in how many would subsequently test positive for amyloid via PET.
Plasma Eligible? Measured by the same blood test, different ethnic and racial groups had different rates of amyloid positivity (orange, eligible; grey, ineligible). [Courtesy of Doris Molina-Henry, University of Southern California, San Diego.]
These findings show that the plasma thresholds are similarly effective across groups at predicting amyloid-PET positivity, Molina-Henry said. In other words, plasma screening does not disproportionately weed out potential participants from any group. The results also jibe with what others have found in the field: namely, that black, Hispanic, and Asian volunteers are less likely to have amyloid accumulation than their non-Hispanic white counterparts.
To Molina-Henry’s mind, this is striking given the higher prevalence of dementia among black and Hispanic people. Although AHEAD 3-45 enrolled cognitively normal people, 75 percent of those screened had a family history of dementia. Molina-Henry thinks that a higher prevalence of other diseases, including cardiovascular illness and diabetes, could drive this result of amyloid-independent forms of dementia among black and Hispanic communities. She emphasized that these comorbidities might in turn exacerbate amyloid-driven AD. These questions are ripe for study.
To do so, the scientists are enrolling people whose plasma or PET result kept them out of AHEAD3-45 into the Alzheimer Plasma Extension observational study. With 762 in it so far, APEX is 75 percent full, and more than half of the enrollees come from underrepresented racial or ethnic groups. It will track sociodemographic and health factors, as well as cognition and AD biomarkers. Molina-Henry hopes the data will reveal underlying causes of dementia and identify better biomarkers for them.
Another prevention effort, TRAILBLAZER-ALZ3, turned up similar results. In Madrid, Eli Lilly’s Karen Holdridge showed data of people who were screened in the trial, which is evaluating donanemab in cognitively normal people with brain amyloid based on plasma p-tau217. To expand its reach geographically and demographically, TRAILBLAZER-ALZ3 performed decentralized screening—ruling out cognitive impairment over the phone with the modified telephone interview for cognitive status (TICSm) and collecting blood for AD biomarker testing at third-party, often remote, sites. They screened 63,124 people in the U.S. and Japan, of whom 2,196 were randomized to drug or placebo. Of the more than 60,000 screen fails, 81 percent were due to low plasma p-tau217, 5.4 percent due to cognitive impairment.
Consistent with AHEAD 3-45, Holdridge reported greater ethnic and racial diversity among those who were screened than among those who were subsequently enrolled. For example, Hispanic/Latino people made up 13 percent of those who were screened, but only 8.2 percent of those randomized. For people identifying as black or African American, the differences were even greater, dropping from 10.5 to 2.6 percent from screening to enrollment. The opposite trend unfolded for white people, who went from 81.7 percent of the screened population to 93 percent of enrollees. Again, these differences were attributed to higher screen-fail rates among non-whites.
After Holdridge’s talk, Anton Porsteinsson of the University of Rochester, New York, noted that compared to the population screened for AHEAD3-45, where 75 percent had a family history of dementia, only 54 percent of participants screened for TRAILBLAZER-ALZ3 reported a first-degree relative with dementia. Similarly, while more than 70 percent of AHEAD3-45 participants were ApoE4 carriers, just over half of enrollees in TRAILBLAZER-ALZ3 were. Holdridge attributes this to a wider net cast during screening, by way of extensive community outreach with mobile research units and blood draws at health fairs. This made screening less reliant on self-selection. The different AD risk profiles of the two participant cohorts in these two prevention trials will make for a good comparison, Porsteinsson said.
Offering a potential counterpoint from a third population, Michael Hodsdon of Eli Lilly presented blood biomarker data from more than 2,000 people who were screened for the TRAILBLAZER-ALZ2 trial of donanemab in people with amyloid and mild cognitive impairment or mild dementia. While primarily about clinical validation of Lilly’s SPX p-tau217 assay, Hodsdon’s talk included a comparison of the assay’s performance across ethnoracial groups.
Nearly two-thirds of screened people were amyloid-positive according to the p-tau217 test, which in turn was more than 90 percent concordant with amyloid-PET, Hodsdon reported. At 65 and 61 percent, respectively, amyloid prevalence was similar between whites and non-whites. Hodsdon did not break down these racial groups by ethnicity, so both the white and non-white groups comprised Hispanic and non-Hispanic people. Likewise, Hodsdon did not report Aβ status for black/African Americans and Asians. In a separate comparison by ethnicity, Hodsdon found that only 37 percent of Hispanic people were amyloid-positive, compared to 80 percent of non-Hispanics. One clear finding from this cognitively impaired cohort was that Hispanic people had much lower rates of amyloid positivity than their non-Hispanic counterparts.
After Hodsdon’s talk, Charlotte Teunissen of Amsterdam University Medical Center noted that data from several studies are converging on the idea that people from underrepresented groups indeed have a lower prevalence of amyloid positivity. She wondered if there are better ways to identify people with underlying causes of cognitive impairment other than AD prior to trial screening. Others concurred, saying the field can now focus on understanding non-AD factors responsible for the higher rate of cognitive impairment in these groups. “Instead of squeezing them in to fit with what we do know about AD, we need to figure out what’s causing these impairments,” one audience member said.
APEX is attempting to do that. The study attempts to chronicle AD biomarkers along with various comorbidities such as cardiovascular disease, diabetes, and stroke, as well as sociodemographic factors in the development of cognitive impairment. “We think it’s disadvantage in these communities as a whole that makes them have higher rates of comorbidities,” Molina-Henry told Alzforum. Those comorbidities, she believes, influence what kind of dementia might develop.
Some of these social factors took center stage in a talk by Gil Rabinovici, University of California, San Francisco. He presented demographics and amyloid-PET results from New IDEAS. Similar to its predecessor, this study set out to test if amyloid-PET scans added enough value to clinical care to warrant coverage by Medicare. New IDEAS set the goal of enrolling at least 4,000 black/African American or Hispanic/Latino participants, among its target of 7,000 participants.
In Madrid, Rabinovici explained that the study fell short of those goals because it was terminated early. This happened in the wake of a decision in October 2023 by the Centers for Medicare and Medicaid Services to expand coverage for amyloid-PET, obviating the need to demonstrate that amyloid-PET warrants coverage. Rabinovici said they initially decided to continue anyway, using the newly expanded coverage to pay for their participants’ PET scans. Alas, problems arose once people’s scans were denied coverage, particularly by Medicare Advantage plans, which black and Hispanic participants were more likely to carry. Ironically, this recreated a disparity New IDEAS had set out to address, hence the scientists decided to stop, Rabinovici told Alzforum.
Even so, New IDEAS registered more than 6,000 people, of whom 4,845 received an amyloid scan at one of the 111 participating PET facilities. They averaged 75 years of age; 64 percent had MCI, 36 percent dementia. Non-Hispanic whites made up 60 percent of those scanned; black/African Americans and Hispanic/Latinos accounted for 21 and 18 percent, respectively.
While 68 percent of non-Hispanic whites were amyloid-positive, 60 percent of people from black and Hispanic groups were. Why is this difference smaller than in other cohorts? Partial reasons could be that New IDEAS reflects a real-world memory care population, not clinical trial volunteers, and also that more non-Hispanic white people saw dementia specialists and received their scans at earlier stages of impairment than did black or Hispanic volunteers, Rabinovici said.
The study also captured differences in sociodemographic and health variables across ethnoracial groups. On average, black and Hispanic people had less formal education than non-Hispanic whites, and more of them were unmarried and living alone. They were less likely to live in a “prosperous/comfortable” neighborhood, as gauged by area deprivation index, a measure of neighborhood disadvantage. For example, while 65 percent of non-Hispanic whites lived in such neighborhoods, 55 percent of Hispanic/Latinos and 42 percent of black/African Americans did.
The prevalence of comorbidities was also higher among black and Hispanic groups. For example, 73 percent of black and 61 percent of Latino people had hypertension, compared to 54 percent of non-Hispanic whites. Diabetes affected nearly 30 percent of people who were black or Latino, but only 16 percent of non-Hispanic whites. Black people had higher rates of stroke, chronic kidney disease, and cerebrovascular disease as well. How these disparities relate to one another, and to prevalence and types of cognitive impairment that arise across groups, is being investigated in ongoing analyses of New IDEAS data, to be published early next year. Blood biomarkers will be measured among a subset of volunteers who opted to give blood, Rabinovici said.—Jessica Shugart
Some Alzheimer’s Blood Tests Are Racing Toward IVD Certification
Yes, blood tests for Alzheimer’s disease are ready for prime time. So pronounced Suzanne Schindler, Washington University, St. Louis, in a keynote presentation at CTAD 2024 last month in Madrid. In answering a question that has been raised often in the last few years, Schindler said that the field has reached a tipping point, and that highly accurate plasma tests deserve much of the credit.
“Things are moving very quickly. I expect in 2025 we may have FDA-approved blood tests that are reimbursed by payers,” Schindler said. “This would really change the landscape.”
Indeed, Jonathan Schott, Queen Square Institute of Neurology, London, noted that in the U.K., only about 2 percent of people who qualify are tested for CSF markers or a PET scan. He thinks blood tests could bring that number close to 100 percent. “We have the potential now to democratize molecular diagnosis,” Schott said. “I think we have an unbelievable opportunity here.”
A number of laboratory-developed tests (LDT) for blood p-tau217 are already commercially available in the U.S. and Europe, and used in research and some specialty clinical care centers. Current LDTs include those sold by ALZpath Inc., C2N, Eli Lilly Clinical Diagnostics Laboratory, LabCorp, Lucent Diagnostics, Mayo Clinic, and others. However, the U.S. Food and Drug Administration stipulated last May that, by 2028, any new plasma AD diagnostic will need approval. Existing tests are exempt, but many vendors will comply to stay competitive (May 2024 community news).
Which vendors are filing? Fujirebio last September requested approval from the FDA for its plasma p-tau217/Aβ1-42 test. At CTAD, Fujirebio’s ManuVandijck said that following the FDA submission, the company also plans to seek IVD approval from the Competent Authorities for Medical Devices in the EU, and the Pharmaceutical and Medical Devices Agency in Japan.
In Madrid, Margherita Carboni, Roche Diagnostics International, told Alzforum that her company, too, had filed for IVD approval from the FDA for its p-tau181 and p-tau217 assays. As has Fujirebio for its test, Roche received FDA breakthrough device designation in July 2002 for its Elecsys Amyloid Plasma Panel, which measures plasma p-tau181 and ApoE4, and for its plasma p-tau17 last April.
Jeff Tarmy, Beckman Coulter Diagnostics, Brea, California, told Alzforum that his company expects to file for IVD certification in the near future. Startup company ALZpath Inc., Carlsbad, California, licensed its p-tau217 antibody to Beckman last July for use in blood-based in vitro diagnostics. Scientists who spoke to Alzforum said they expect other vendors to file soon, as well. Those in the running include C2N Diagnostic’s mass spectrometry-based test for the p-tau217/tau217 ratio, Eli Lilly’s p217 immunoassay, and others.
Leaders of AD drug development programs told Alzforum they are urgently awaiting regulatory approval for those tests, especially in Europe, where IVD status is often required for a plasma test to be used in investigational drug trials. In the U.S., laboratory-developed tests can be used in clinical trials.
How have things evolved so quickly? One driving force was diagnostic criteria issued by a Global CEO Initiative panel. Led by Schindler and Oskar Hansson from Lund University, Sweden, the panel earlier this year recommended that for AD triage in primary care, blood biomarker tests have a sensitivity and specificity of at least 90 and 85 percent, respectively, and that two separate cut points be used to identify people who are positive, negative, or indeterminate. Spurred by this, vendors are now optimizing their tests to meet these criteria.
Ratio Works Best. Vendors want their tests to return as few “indeterminate” results as possible. For Fujirebio, this means using the p-tau217/Aβ1-42 ratio. [Courtesy of Manu Vandijck, Fujirebio.]
As with every test, the positive and negative predictive value of a p-tau217 test, PPV and NPV for short, will depend in part on the prevalence of the tested condition in the population being screened (discussed in Aug 2024 conference news). Hence, vendors are validating their tests in various clinical settings.
In Spain, Francesca De Simone, Fujirebio Diagnostics Inc., Malvern, Pennsylvania, reported that, in their hands, the p-tau217/Aβ1-42 ratio outperforms p-tau217 alone. In samples from 208 volunteers—44 from the Swedish BioFinder-2 study, 66 from the BIOCARD study at Johns Hopkins University, and 98 from the MissionAD trial of a discontinued BACE inhibitor—p-tau217 identified amyloid-positive people with PPVs and NPVs of 92 and 93 percent, respectively, which is within the CEOi criteria. However, for 34.2 percent of them the test was indeterminate. The CEOi panel recommends that no more than 20 percent of people fall into this gray zone.
At CTAD, De Simone showed that the p-tau217/Aβ1-42 ratio did better in this regard. The PPV and NPV reached 95 and 96 percent, respectively, and only 20 percent of samples were indeterminate. These results highlight a high performance of the p-tau217/Aβ1-42 ratio in predicting amyloid positivity, De Simone said. Vandijck reported finding the same numbers in a separate cohort of 500 volunteers. Twenty percent fell into the gray zone using the ratio, while 34 percent were indeterminate based on p-tau217 alone.
This held up in analysis of plasma samples in the Phase 3 AD trial of AriBio Inc.’s phosphodiesterase inhibitor AR1001. Sharon Sha, Stanford University, California, reported that for 256 samples the PPV and NPV for p-tau217/Aβ1-42 were 91 and 97 percent, respectively, with 21 percent indeterminate. For p-tau217 alone, PPV and NPV were comparable, 37 percent indeterminate.
For its part, Roche seems to be sticking with p-tau181+ApoE4 and p-tau217 alone. In Madrid, Carboni reported that the latter performs well across the AD spectrum. One burning question in the field is how well plasma markers will work in different populations, especially those where prevalence of AD may be low. Roche has tested its p-tau217 assay across five cohorts comprising people who are 60 and older. They span the AD spectrum from cognitively normal and an amyloid prevalence of 11 percent, to those clinically diagnosed with dementia, where the prevalence runs close to 60 percent. Carboni did not report PPVs and NPVs but said the AUCs, a measure of sensitivity and specificity, was 0.88 or higher across the spectrum.
“These evaluations have demonstrated robust clinical and technical performance across all cohort,” Carboni told the audience. Roche plans to test this assay across broader populations to prove it is useful and reliable. Carboni said they will use the two-cutoff method and are working to get the number of indeterminate reads to below 20 percent.
P-tau217 Alone? Eli Lilly’s CertuitAD relegates only 18 percent of samples into the indeterminate zone. [Courtesy of Eli Lilly Clinical Diagnostics Laboratory.]
Lilly scientists have also settled on analysis of p-tau217 alone for their CertuitAD test, which is available commercially in the U.S. as an LDT for clinical diagnosis. It uses Lilly antibodies and runs on the Quanterix imaging system. Lilly was first to establish a plasma assay for p-tau217, based on antibodies developed by Jeff Dage, who is now at Indiana University, Indianapolis.
In Madrid, Lilly’s Rose Beck reported that among 2,071 volunteers who had been screened for the Trailblazer-Alz2 trial of donanemab, CertuitAD identified amyloid positives with a PPV and an NPV of 95 and 84 percent, respectively. Only 18 percent of this cohort, who were 60 and older with cognitive complaints, were indeterminate. On this basis, this assay would meet the CEOi criteria for an IVD. Beck told Alzforum that it is not clear if Lilly will file for approval with the FDA.
Post Approval
What happens once IVD blood diagnostics are approved? Scientists at CTAD agreed that work needs to be done to integrate them into clinical practice. “We need many more real-life studies to see how these tests perform in diverse settings,” said Schott during a roundtable discussion led by Marc Suárez-Calvet, Barcelonaβeta Research Center, Spain.
Researchers around the world are stepping up. In South Korea, scientists led by San Won Seo at Samsung Medical Center, Seoul, asked if having different high and low cutoffs for plasma p-tau217 at different stages of the Alzheimer’s helps improve test performance in their country. They tested samples from 2,699 participants in the Korea-Registries to Overcome Dementia and Accelerate Dementia Research. Part of the worldwide ADNI program, K-ROAD has recruited more than 5,800 people, between 63 and 80 years old, across 28 centers (Jang et al., 2024).
In Madrid, Jehyun Ahn, also at Samsung Medical Center, showed that among cognitively unimpaired people aged 65 and younger, lowering both cutoffs improved both PPV and NPV. For those over 65, only the NPV improved by using a group-specific cutoff. Among people with mild cognitive impairment or with a clinical diagnosis of AD, group-specific cutoffs did not improve diagnosis. Ahn did not report data on how other factors, including sex, ApoE genotype, comorbidities, or ethnicity might affect these cutoffs, but thinks this should be investigated.
Others are investigating how plasma tests will change clinical practice, much like IDEAS in the U.S. and AMYPAD in Europe assessed how amyloid PET scans influence disease management (April 2019 news; Sep 2022 conference news). Lilly’s clinical utility study for CertuitAD is testing how plasma p-tau217 results affect diagnosis, treatment, and management; it will read out in early 2025, said Beck. In the U.K., Schott is planning a randomized trial to test how disclosing blood test data three and 12 months later affects diagnosis and patient outcomes. “We have learned a lot about how to improve utility from IDEAS and AMYPAD. Touch wood, this should be easier with blood tests than with PET,” said Schott. Michael Schöll, University of Gothenburg, is running a similar study in southwestern Sweden (Jan 2024 news).
Scientists agree that these tests should only be used as part of a broader clinical evaluation. “There is a danger that a positive test will be seen as a clinical diagnosis,” warned Schott. An important aspect will be education for clinicians who may be naïve about molecular diagnoses. Suárez-Calvet, whose center uses blood-based biomarkers for both research and to support diagnosis, said that rolling the tests out with abundant physician education is important to prevent false use and a possible backlash. Marwan Sabbagh, Barrow Neurological Institute, Phoenix, agreed that physicians will need guidance. “I would start using these tests for their negative predictive value, not positive, and I would only use them in patients who have memory complaint,” Sabbagh said.
Appropriate-use recommendations for these tests will be issued. The Alzheimer’s Association is leading an effort to do that. “We start with clinical practice guidelines for specialty care,” said the association’s Rebecca Edelmayer. They will be based on current evidence, which at this point comes from secondary care settings. “We need to drive toward primary care, but that will require an enormous amount of clinical utility research,” Edelmayer said. The guidelines will evolve into a collection to support the right tests for the right patients at the right time, Edelmayer said
Who will pay for these tests? Dage said that the Centers for Medicare and Medicaid Services was proposing to reimburse at a rate of $17.27 per test. At that rate, labs will lose money and won’t implement the tests, Dage wrote to Alzforum. How did the CMS come up with that rate? “It is called cross-walking to an existing test,” according to Dage. Generic immunoassays are currently reimbursed at that rate, and the CMS extrapolates to AD.
CSF tests for some of those same analytes are reimbursed at $130. “Given the complexity of the blood tests, including the need for ultrasensitive detection and specialty high-affinity reagents, they should be reimbursed at a minimum of the $130 rate,” wrote Dage. The deadline to comment on the proposed fee passed on October 25. At time of writing, the final CMS payment schedule had not been posted.—Tom Fagan
Beyond Antibodies: From CTAD, New Attempts at Outflanking Alzheimer’s
Most Alzheimerologists agree that lecanemab and donanemab are a start, but insufficient to treat this disease. At the Clinical Trials in Alzheimer’s Disease conference, held last month in Madrid, scientists not only debated how to make anti-Aβ monoclonals safer and more effective (Part 2 of this series; Part 1 of this series; Part 6 of this series), but also shared findings from Phase 1 trials evaluating new approaches. Gene silencing looked like it might shush APP and tau, and a gene therapy cranked up the protective ApoE2 protein while possibly nudging AD biomarkers. Arrows aimed at tau—an antisense oligonucleotide and a monoclonal antibody—might take down tangles. Meanwhile, immune checkpoint inhibitors and intranasal insulin are budding to target other aspects of the disease.
In her keynote presentation in Madrid, Catherine Mummery of University College London showed an image of a picturesque valley with the snowy peak of Nanga Parbat looming in the distance. Mummery noted the progress the field has seen over the past few years in its approach. “We are in the foothills of our treatment journey,” she said, “If we want to get to those dizzying heights of cures, or at least of halting the disease, then we need to redouble our efforts.”
Looong Trek to Summit. At CTAD, Nanga Parbat served as inspiration for trialists working to build a therapeutic approach to conquering an imposing, scary disease.
Down With APP
For starters, how else can scientists thwart amyloidosis? One approach is mivelsiran, previously known as ALN-APP. Developed by Alnylam of Cambridge, Massachusetts, with Regeneron in Tarrytown, New York, this small interfering RNA targets the APP transcript for destruction. Conjugated to a lipid chain that greases its passage into cells, this siRNA takes down APP and all its downstream metabolites, Aβ42 and Aβ40 among them.
Mivelsiran is in Phase 1 in AD, and in Phase 2 for cerebral amyloid angiopathy. Previously, Mummery had presented interim safety and biomarker data from a single-ascending-dose trial in people with early onset MCI or mild AD, who received an intrathecal infusion of placebo or 25 mg, 35 mg, 50 mg, or 75 mg of mivelsiran. A 70 percent drop in APP metabolites in CSF on the two higher doses stayed 30 percent below baseline by six months (Nov 2023 conference news).
In Madrid, Mummery reported 12-month findings for the two highest dose groups. No ARIA occurred, though adverse events related to lumbar puncture did. On both 50 mg and 75 mg, CSF sAPPb remained 30 percent below baseline. Mummery also showed six-month data for Aβ40 and Aβ42, in which the 75 mg group remained 27 and 40 percent reduced relative to baseline, respectively. This finding was not dose-dependent, however.
Durable Effect. Metabolites APPβ (top), Aβ40 and Aβ42 (bottom) plummeted after treatment with 35, 50, or 75 mg mivelsiran. [Courtesy of Alnylam Pharmaceuticals.]
According to Alnylam’s Tim Mooney, mivelsiran’s endurance is due to stabilizing tweaks the scientists made to the siRNA backbone. Once the construct enters cells, it can repeatedly silence mRNAs before it gets degraded. “It can punch above its weight,” Mooney said, adding that future work will define the required dosing frequency.
Both this study, as well as multi-dose studies, are ongoing among people with EOAD. Mummery considers mivelsiran’s potential for infrequent dosing a plus. After all, who wants monthly infusions near their spinal cord? “Giving such a drug once every nine to 12 months is a different prospect,” she told Alzforum.
Importantly, mivelsiran is also being evaluated to treat CAA. Not only is this cerebrovascular condition a common menace in its own right, but it often co-occurs in people with AD (Jäkel et al., 2021). CAA is difficult to diagnose but people with manifestations of it, such as superficial siderosis and microbleeds, are at increased risk for hemorrhage while on anti-amyloid antibodies. Both people with CAA alone, or with both conditions, need disease-modifying treatments. Could mivelsiran fit the bill?
In Madrid, Jin-Moo Lee, a stroke specialist who heads the neurology department of Washington University, St. Louis, laid out the rationale and design of the ongoing Phase 2 trial. Called cAPPricorn-1, it is enrolling people diagnosed with sporadic CAA as gauged by an updated version of the Boston criteria, which use several MRI features to infer cerebrovascular signs of CAA (Charidimou et al., 2022). People with MCI or mild AD in addition to CAA are eligible, though people with moderate or severe AD are not.
A separate “descriptive” cohort, which won’t count toward the primary endpoints, will enroll people with Dutch-type CAA. This rare, familial form of the disease worsens rapidly. It is caused by the E693Q APP mutation, located in the Aβ sequence itself. Its carriers typically suffer from recurrent strokes starting in midlife; some are members of DIAN.
Participants in this trial will receive a single intrathecal infusion of mivelsiran or placebo, followed by a two-year double-blind follow-up period. The primary endpoint is change in how many microbleeds arise in the cerebral lobes. Secondary endpoints include hemorrhagic and cerebrovascular signs of disease progression, most measured by MRI. Change in CSF sAPPα and sAPPβ is on the list, as are other fluid and imaging biomarkers designated exploratory endpoints, Mooney told Alzforum.
Depending on how both trials go, mivelsiran could be tried in late-onset AD, for example in ApoE4 homozygotes, or people who take medications that up their ARIA risk in response to anti-Aβ antibodies. Mummery suspects mivelsiran will work best to prevent amyloidosis, as it turns off the spigot of production.
Down With Tau
Only in the earliest stages of Alzheimer’s, years before symptoms, do anti-amyloid therapies stand a chance of stopping this disease in its tracks. For most people, halting progression will require hitting other parts of the cascade, likely more than one. “We need to target different modes of action if we want to make a greater difference beyond just nudging the needle,” Mummery told Alzforum.
Notable here is recent progress in targeting tau, which starts to misbehave earlier in AD progression than previously thought. A highlight at CTAD was a promising signal for bepranemab, the first tau immunotherapy that appears to remove tangles (Nov 2024 conference news).
Until then, the lone tau drug to have done that was Biogen’s antisense oligonucleotide BIIB080. It dampens tau expression and, in a Phase 1 trial, reduced tangles as per tau-PET (Apr 2023 conference news; Nov 2023 conference news). At CTAD, Mummery presented tau-PET trajectories of individual participants in this trial. Across Braak regions, nearly every participant saw their tau accumulation slow over the course of the trial. BIIB080 is now being evaluated in a Phase 2 trial, which is fully enrolled. Meanwhile, Eli Lilly recently threw its hat in the ring, starting a with an siRNA against tau called LY3954068.
Down with ApoE4
As the strongest AD risk gene, ApoE makes for a tantalizing therapeutic target. One approach attempts to reduce the negative effects of the high-risk ApoE4 isoform by forcing expression of the protective isoform, ApoE2. This is the rationale behind Lexeo’s LX1001, an adeno-associated virus (AAV) carrying the ApoE2 gene. At CTAD, Kim Johnson, a site investigator at Duke University in Durham, North Carolina, showed interim findings from an ongoing Phase 1/2 trial evaluating LX1001 among homozygous ApoE4 carriers with MCI or mild to moderate AD.
The 15 participants are divided into four little cohorts of three to five people, who received a single ascending dose of LX1001, ranging from 5.7 x 1012 vector genomes for cohort 1, to 1.4 x 1014 vector genomes for cohort 4. The drug entered their CNS via intrathecal injection between their top two cervical vertebrae. At a previous CTAD, interim data from five participants in the two lowest dose groups looked as if the treatment safely stoked CSF ApoE2 production (Dec 2022 conference news). In Madrid, Johnson reported 12-month follow-up data for cohorts 1 to 3, and six-month data from cohort 4.
By this point, LX1001 was still largely safe and well-tolerated across cohorts, and no one developed ARIA, Johnson said. Twelve participants developed CSF pleocytosis, aka elevated white blood cells. This condition has been reported in response to other AAV therapies. It peaked at three months, receded by six months, and remained at bay at 12 months. Four serious adverse events cropped up during the study, of which one—mild hearing loss—was deemed potentially related to the drug. Johnson told Alzforum that the participant still has hearing loss, which is mild enough to be corrected with hearing aids.
Three months after injection, CSF ApoE2 ramped up in a LX1001 dose-dependent manner, holding steady at six and 12 months. CSF ApoE4 remained stable at the lower doses, but appeared to rise in cohort 4. The CSF ratio of ApoE2 to ApoE4 rose at three months, stabilizing thereafter (image below). CSF ApoE2 skyrocketed in one participant from cohort 3 at three and six months. This outlier declined to undergo lumbar puncture or PET at 12 months.
ApoE2 to the Rescue? CSF ApoE2 (left) rose three months after injection, stabilizing later. CSF ApoE4 stayed the same on the lower doses (middle). The CSF ApoE2/ApoE4 ratio increased dose-dependently (right). [Courtesy of Kim Johnson, Duke University.]
Amyloid-PET showed no significant change in amyloid accumulation. This measure was too variable to say whether the drug had any impact in such a small sample size, Johnson said.
What about tau biomarkers? For nine of the 13 participants, CSF p-tau181 and total tau had decreased relative to baseline by their last follow-up visit. P-tau217 and p-tau231 were measured in cohorts 2 and 3, where both dropped relative to baseline in four of the six participants.
Tau-PET scans at baseline and six months for cohorts 3 and 4, and out to 12 months for cohort 3, suggested that tau deposition stabilized, and for some, appeared to go down. The outlier with highest CSF ApoE2 had the steepest tau-PET reduction at six months.
Gil Rabinovici, University of California, San Francisco, asked if the tau-PET scans had corrected for volume loss, to ensure the findings weren’t an artifact of ongoing brain shrinkage. Alzforum asked the company the same question, and Lexeo's Sandy See Tai clarified that Lexeo does not perform partial volume correction. "However, we use concurrent MRIs to each PET scan to define the target regions of interest for the PET SUVR analysis, therefore, atrophy that has occurred since baseline is taken into account," See Tai wrote to Alzforum.
Johnson acknowledged that, despite the encouraging CSF biomarker and tau-PET results, the study is too small to draw conclusions. Complete data from the 52-week trial will be available soon, and participants will enter a long-term extension to measure CSF ApoE2, CSF biomarkers, and amyloid- and tau-PET annually for four years.
Even as LX1001 moves through its trial, a new mouse study by Lexeo’s founder, Ronald Crystal of Weill Cornell Medical College in New York, claims that adding a protective Christchurch mutation to the AAV-packaged ApoE2 might strengthen the protection (Günaydin et al., 2024).
Wrangling Inflammation and Metabolism
Dysfunction in these two areas is well recognized in AD. Tweaking inflammation is dicey, as scientists are still exploring which immune responses are beneficial versus detrimental. What’s more, immune responses change as disease progresses.
One controversial approach seeks to take the breaks off the immune system with checkpoint inhibitors such as antibodies against programmed cell death-1 receptor and its ligands PD-L1 and PD-L2. Preclinical work from Michal Schwartz’s lab at the Weizmann Institute of Science in Rehovot, Israel, supports this approach (Jan 2016 news; Feb 2019 news). To carry it into humans, Schwartz co-founded ImmunoBrain Checkpoint, which is running early dosing studies of an anti-PD-L1 antibody in people with early AD, with Mummery as its principal investigator.
The studies are testing six doses of an anti-PDL1 antibody in 40 people with early AD. They receive an intravenous infusion of placebo or the antibody once every quarter for 12 months. In Madrid, Mummery said that administration of the fifth dose is happening now. As expected for a checkpoint inhibitor, there have been mild immune-related adverse events, but no worrying safety signals so far, she said.
Also at CTAD, Suzanne Craft of Wake Forest School of Medicine in North Carolina reported biomarker data from a one-month trial of intranasal insulin alone or in combination with empagliflozin. Insulin exerts metabolic and immune effects in the brain; “Empa” is a sodium-glucose co-transporter 2 (SGLT2) inhibitor that reduces glucose uptake and improves vascular function. Craft hopes that the two drugs together may act additively or synergistically. She reported that a new intranasal device, called Apgar CPS, delivered insulin more effectively into the brain than was seen in a previous trial; radiolabeled insulin-PET scans are used to confirm exposure.
The month-long trial included 47 people with a clinical diagnosis of MCI or early AD, who were randomized to placebo, 40 IU of intranasal insulin four times per day, 10 mg of empa per day, or both. At baseline and four weeks, CSF and plasma biomarkers, MRI, and cognition were measured. No treatment-related adverse events cropped up.
A few possible signals from this trial: exploratory findings included improved scores on the PACC5, and more intact white matter among people on intranasal insulin alone or with empa. People on empa—with or without insulin—had a drop in CSF total tau. Craft showed changes in CSF and plasma markers of inflammation, as measured by Alamar’s Nulisa platform. In CSF, insulin stoked brain-derived neurotrophic factor while quelling the synapse-targeting complement protein C1qa. Empa reduced pro-inflammatory proteins including IL-6 and TNFα.
Surprisingly to Craft, both treatments nudged plasma markers. Intranasal insulin reduced pro-inflammatory proteins such as interferons, chemokines, and adaptive immune regulators, whereas Empa docked plasma levels of the immune regulators NAMPT and SIRPa, both of which have been implicated in AD risk. These effects were strongest among those with higher baseline p-tau217, suggesting people with amyloid are likely to benefit from the treatment.
These responses could be due to chance in this small sample. Or they could reflect immune communication between the CNS and periphery via the nasal olfactory plexus, Craft believes. Positioned behind the nasal cavity, this structure is an immune hub and part of the glymphatic system (for review, see Chae et al., 2024). Craft hopes her approach will find use alone or with anti-Aβ immunotherapy.
For her part, Mummery envisions combination therapies that address multiple mechanisms contributing to AD. Each therapy might be deployed for different stages of disease, defined by the field’s ever-growing arsenal of biomarkers.
Working toward this future, Mummery heads the U.K.’s Dementia Translational Research Collaboration (D-TRC), a government-funded initiative to create a network of clinical trial sites across the U.K. (Feb 2024 community news). Sites in this network will receive training and resources to run Phase 1 and 2 trials for innovative therapies, including gene therapies that require a deep technical expertise. Mummery told Alzforum that so far, 30 sites have applied to join the network. Companies interested in running early stage trials in the U.K. can tap the network to access sites equipped with the facilities to run such studies. In Madrid, D-TRC investigators got together with interested biotech partners to hash out plans for future trials.—Jessica Shugart
What if there were a way to triple the speed of MRI scans without losing resolution? At the Clinical Trials on Alzheimer’s Disease conference, held October 29 to November 1 in Madrid, Miguel Rosa-Grilo of University College London presented just such an option. By leveraging recent advances in scanning technology, his group cut the time needed for diagnostic structural MRIs by two-thirds, with no measurable loss of quality. Shown side-by-side comparisons of scans done by the new versus old methods, scientists in Madrid could not tell the difference. Adopting the new method could make scanning easier on patients and help remove a bottleneck in Alzheimer’s diagnosis and care, Rosa-Grilo suggested.
Clifford Jack of the Mayo Clinic in Rochester, Minnesota, agreed the field needs to move in this direction. He noted that fast MRI scans using a similar protocol are being tested in ADNI4 and the Standardized Centralized Alzheimer’s & Related Dementias Neuroimaging (SCAN) study, as well.
“Speeding up MR exams will be necessary to accommodate increased throughput as a new stream of patients receiving AD treatment places increasing demands on radiology departments,” Jack wrote to Alzforum (comment below).
Spot the Difference? T1-weighted scans of a 59-year-old with posterior cortical atrophy, done by standard and fast protocols. Which is which? (Answer at bottom.) [Courtesy of Miguel Rosa-Grilo.]
In Alzheimer’s, MRI scans are needed not only for diagnosis, but also to monitor for ARIA, the most concerning side effect of amyloid immunotherapy. The large number of required scans can limit how many patients a site can treat. In addition, long scan times can be hard on patients. Some have trouble tolerating the claustrophobia of the machine or lying motionless for long.
To speed things up, Rosa-Grilo, working with Nick Fox at UCL, turned to Wave-Controlled Aliasing in Parallel Imaging. Developed in 2015 by scientists at Massachusetts General Hospital, Wave-CAIPI uses multiple coils arranged around a patient’s head to perform parallel imaging in three-dimensional space, speeding up acquisition (Bilgic et al., 2015).
Rosa-Grilo incorporated Wave-CAIPI into the diagnostic scanning protocol used at UCL’s memory clinic. It comprises four sequences: T1-weighted 3D, T2-weighted 2D, T2-weighted 3D, and 3D FLAIR imaging. The entire protocol takes nearly 18 minutes to complete. By adding Wave-CAIPI, Rosa-Grilo shortened this to 6.5 minutes.
A FLAIR for Speed. Three-dimensional FLAIR imaging of a 74-year-old with vascular cognitive impairment, done by both protocols. Which is which? (Answer at bottom.) [Courtesy of Miguel Rosa-Grilo.]
Was the faster imaging as reliable? To test this, the scientists recruited 90 patients who came to the clinic for diagnosis, scanning them with both the old and new protocols in the same session. In this group, 21 percent turned out to have AD, 38 percent other pathologies, and 41 percent no pathology. Three neuroradiologists independently assessed their scans in a blinded fashion.
The result? The radiologists assessed the fast and standard scans of each patient nearly identically on multiple measures, including detecting the presence of medial temporal lobe atrophy, posterior cortical atrophy, microbleeds, and white-matter hyperintensities. The type of scan made no difference in how likely radiologists were to clear AD patients for amyloid immunotherapy. Moreover, the variability between how a given rater assessed a fast versus standard scan of the same patient was smaller than the variability between how different raters assessed those scans. Because use of the fast scan introduced less variability than did changing raters, fast scans are no worse than standard ones, Rosa-Grilo concluded.
Finding Microhemorrhages. Susceptibility-weighted imaging to detect microbleeds (arrows) in a 60-year-old with cerebral amyloid angiopathy, done by both protocols. Which is which? (Answer at bottom.) [Courtesy of Miguel Rosa-Grilo.]
To illustrate his point, Rosa-Grilo showed several examples of fast versus standard scans of the same patient, and asked the audience to guess which was which. In general, the audience was unable to tell. Can you do better?
The findings suggest that the fast protocol can be used for diagnosing AD and assessing treatment eligibility, Rosa-Grilo said. He noted that it remains to be shown whether the fast protocol will work for monitoring ARIA, as well. Jack, for one, believes it will. “Although ARIA was not specifically evaluated, the imaging exam described by Rosa-Grilo and colleagues includes both FLAIR and gradient echo sequences and thus should be useful for detection of ARIA,” he wrote. Rosa-Grilo is now testing this.
Other scientists agreed the advance has potential. Some noted that fast scans may save less time in routine clinical care than they did in this study, because of the required check-in, undressing, and redressing time each patient needs for these appointments. Still, Jonathan Schott of Queen Square Institute of Neurology, London, who works with Fox and Rosa-Grilo, estimated that the new technology might bring the current 30-minute time slots their clinic now schedules for diagnostic scans down to 15 minutes.
Schott noted another advantage: Because scanning is so rapid, it would be possible to quickly redo the sequence if the patient moved during the procedure. That would help scientists capture better data.—Madolyn Bowman Rogers
Answers: 1. Fast scan on left. 2. Fast scan on right. 3. Fast scan on right.
In Familial Alzheimer’s, β-Synuclein Rises in Blood a Decade Before Onset
Alzheimer’s disease is a long, slow neurodegenerative calamity, and scientists are still hunting for blood biomarkers that reflect all its specific stages. At the Clinical Trials on Alzheimer’s Disease meeting, held last month in Madrid, β-synuclein emerged as a candidate indicator of early synaptic struggles.
Not to be confused with its infamous cousin α-synuclein, β-synuclein has no relationship to the Lewy bodies found in Parkinsonian disorders. However, it may have something to do with AD. Patrick Oeckl of the German Center for Neurodegenerative Diseases in Ulm reported that β-synuclein starts to rise in the blood more than a decade before the first flicker of cognitive symptoms arise in people who carry a dominantly inherited AD mutation. It changes after biomarkers of amyloidosis, but prior to indicators of axonal damage, suggesting it may help fill a current gap in staging AD.
β-synuclein is a presynaptic protein that is only expressed in the brain. Previously, Oeckl and colleagues had reported that its levels were down in brain homogenate from people who had died with sporadic AD (Halbgebauer et al., 2020). Later, the scientists found that β-synuclein started creeping upward in both plasma and CSF in the preclinical stages of LOAD (Barba et al., 2023; Oeckl et al., 2023). It continued to rise as AD progressed, and correlated with multiple AD biomarkers and cognitive decline.
To nail down when, and perhaps why, blood β-synuclein rises in presymptomatic AD, Oeckl and colleagues turned to the Dominantly Inherited Alzheimer’s Network. DIAN is a standing cohort that includes families with pathogenic APP and presenilin mutations from around the world, including Germany. The scientists used a validated mass-spectrometry-based technique to measure β-synuclein in blood samples from 69 noncarriers, 78 asymptomatic, and 31 symptomatic mutation carriers (Oeckl et al., 2020).
Why does Oeckl use serum? Simply put: Because he can. Some blood markers work only in plasma, creating much demand on increasingly scarce aliquots from unique longitudinal samples from cohorts such as DIAN. By contrast, serum is relatively plentiful and easier to obtain for research, Markus Otto of Martin Luther University Halle-Wittenberg told Alzforum, adding that the β-synuclein assay works in plasma, as well.
β-Synuclein on the Rise. Serum β-synuclein was higher among mutation carriers relative to noncarriers, and highest among those with symptoms. [Courtesy of Patrick Oeckl, DZNE Ulm]
Serum β-synuclein concentrations, which tend to be in the low picogram/ml range in human blood, were higher among asymptomatic mutation carriers than noncarriers, and shot up several fold among carriers who already had memory loss. Using the average age at symptom onset unique to each participants’ mutation, the scientists charted serum β-synuclein levels with respect to expected years to onset (EYO). They found that serum β-synuclein started to rise around 11 years prior to a person’s expected onset, and continued to increase out to 15 years thereafter (image at right).
With respect to symptom onset, β-synuclein started to rise in blood around the same time as did p-tau205 and total tau in CSF. Its change came after biomarkers of amyloid accumulation, including CSF Aβ42/40, but before serum NfL (image below). Among the growing number of tau markers scientists are studying, β-synuclein’s fellow traveler, p-tau205, is considered an intermediate-stage one. It changes after p-tau217, p-tau181, and p-tau231, which indicate that brain amyloid is starting to affect tau processing, but before the later marker MTBR-243, which is a marker of aggregated filaments and tangles.
A Place in the Pack. Serum β-synuclein (red) changes after markers of amyloidosis such as CSF Aβ42/40 (orange), but before serum NfL (blue). [Courtesy of Patrick Oeckl, DZNE, Ulm.]
In keeping with this, Oeckl reported that serum β-synuclein started its ascent after cortical PiB-PET scans became positive, but before FDG-PET scans indicated flagging glucose metabolism and MRIs indicated brain atrophy.
Also fitting in, he reported that β-synuclein predicted amyloid-PET positivity with 75 percent accuracy—a far cry from the 99 and 95 percent predictive value of p-tau217 and p-tau181, respectively. “This means that β-synuclein is only an indirect marker of amyloid pathology,” Oeckl said.
β-synuclein was most strongly associated with markers of neurodegeneration, including waning glucose metabolism, hippocampal shrinkage and, most strongly, cognitive impairment. Its serum concentration rose sharply as scores worsened on the MMSE or the CDR-SB.
What is going on? Oeckl thinks β-synuclein might spill out when synapses degenerate, casting it as an early marker of synaptic problems. Oskar Hansson of Lund University offered an alternative hypothesis. He suggested that β-synuclein could be released due to neuronal hyperactivity, which is known to occur in response to Aβ. Hansson, along with Oeckl, previously reported that β-synuclein rises modestly in sporadic AD. He said it was interesting that the marker appears to rise more steeply among those with autosomal-dominant forms of AD.
Other fluid biomarkers, such as neurogranin, SNAP-25, and neuronal pentraxin, have also been tied to synaptic woes. How does β-synuclein fit in, and what does it add? Oeckl told Alzforum that while he did not measure these other synaptic markers in samples from the DIAN cohort, his previous studies in people with sporadic AD suggest that β-synuclein rises in concert with these other synaptic markers, suggesting related mechanisms. That said, β-synuclein has one advantage, Oeckl told Alzforum: “It is so far the only synaptic marker that can be reliably measured in blood.” Not only is it detectable there, but its sole expression in the brain rules out interference from peripheral sources. He noted that neurogranin, for instance, is also expressed in peripheral tissues, while SNAP-25 is difficult to measure in blood.
How might this new blood marker be used? Oeckl said that in preclinical AD, it could signify the coming onset of cognitive impairment, and therefore suggest a possible time to start treatment. It might also come in handy for trial screening, or to monitor responses to treatment, he added. The scientists are characterizing β-synuclein in other neurological diseases, and evaluating its potential for clinical use. For now, they are optimizing their mass spec-based assay, which avoids the potential for cross-reactivity with α-synuclein that can happen with antibody-based tests.
On the Rise? Whether using an antibody specific for β-synuclein’s N- or C-terminus, an ELISA detected an uptick across clinical stages of AD, though with extensive overlap between groups. [Courtesy of Eugeen Vanmechelen, ADx Neurosciences.]
Speaking of immunoassays, Eugeen Vanmechelen of ADx Neurosciences in Gent, Belgium, at CTAD showed findings from one to detect β-synuclein. Using an ELISA with antibodies specific for the protein’s N- or C-terminus, they detected an uptick in CSF β-synuclein with worsening clinical stages of AD among 160 people from the Amsterdam Dementia Cohort (image below). To detect β-synuclein in serum or plasma, Vanmechelen and colleagues are validating their antibody-based assays on the Simoa platform.—Jessica Shugart
'Presymptomatic AD' or 'Asymptomatic at Risk'? Dx Criteria Disagree
As diagnostic criteria for Alzheimer’s disease are being refined, the U.S. and Europe have taken different paths. Broadly speaking, leading clinicians in the U.S. use Alzheimer’s Association criteria, while those in Europe hew to recommendations from the International Working Group. Both sides agree AD should be defined as a biological disease based on its pathology. Both have been slowly working toward this goal for nearly 20 years, issuing periodic iterations of updated criteria that incorporate new biological evidence every time.
Where they still differ is in how to label cognitively unimpaired people who have biomarker evidence of AD. The Association’s criteria consider these people to have preclinical AD, full stop. The IWG approach calls them “asymptomatic at-risk.” Both groups have some members from the “other” continent, as well.
At the Clinical Trials on Alzheimer’s Disease conference, held last month in Madrid, Howard Feldman of the University of California, San Diego, laid out IWG’s reasoning. Because many older people with amyloid plaques in their brains will not develop symptoms of AD, diagnosing them as such could cause them unnecessary distress and use up scarce medical resources, Feldman said. Labeling such people as at-risk instead would enable clinicians to discuss preventative measures with them, without pathologizing their condition. “We’re providing an alternative viewpoint to that of the Alzheimer’s Association working group,” he noted.
Some scientists in Madrid pushed back on this approach, arguing that people with Alzheimer’s pathology already have the biological disease. They noted that because current Association criteria do not recommend biomarker testing in people without symptoms, defining biomarker positivity as Alzheimer’s disease does not affect clinical practice, but is important conceptually.
Both viewpoints were featured in the November 1 JAMA Neurology, in articles penned by Bruno Dubois of University Salpêtrière Hospital in Paris and colleagues, and Ron Petersen of the Mayo Clinic in Rochester, Minnesota, and colleagues, respectively. Petersen told Alzforum he sees the difference between the positions as largely semantic at this point, since the groups agree on patient care.
Patients in Waiting? Worldwide, only a quarter of people with amyloid plaques are estimated to be cognitively impaired (medium and dark blue). [Courtesy of Howard Feldman.]
Spearheaded by Dubois and Feldman, the IWG was the first to put AD diagnosis on a biological footing, by recommending bolstering the clinical evaluation with supportive biomarkers (Jul 2007 news). The U.S. National Institute on Aging, with the Alzheimer’s Association, issued its own first stab soon after (Apr 2011 news). It took a further step in 2018, defining the disease solely by biomarkers, though only for research purposes (Apr 2018 news; Nov 2018 conference news). This led IWG to emphasize that AD clinical diagnosis should be restricted to people with symptoms (Dubois et al., 2021).
The latest rewrite of the Association criteria agrees with this approach in practice, but not in principle. It again defines AD as positivity on amyloid and tau biomarkers (Aug 2023 conference news; Nov 2023 conference news). The IWG includes more clinicians who see patients regularly than does the Association’s working group, which includes more clinician-researchers focused on biomarker development.
In Madrid, Feldman argued that biomarkers are imperfect predictors of disease progression. He pointed to a study from Rik Ossenkoppele of Lund University, Sweden, that found that people positive for amyloid, but not tau, had little risk of progressing over the next three years, while those who were A+T+ had a 50 percent risk (Ossenkoppele et al., 2022). The likelihood that an A+T+ person will become cognitively impaired over three years varies widely between studies, ranging from 33 to 88 percent, according to a meta-analysis (Strikwerda-Brown et al., 2022). “Biomarkers are probabilistic, not deterministic,” Feldman concluded. “There must be a clinical context for biomarker interpretation.”
Feldman acknowledged that for certain groups with a very high risk, it would be appropriate to diagnose AD in asymptomatic patients. These include people with an autosomal-dominant gene or Down’s syndrome. They would be labeled as having presymptomatic AD, rather than being asymptomatic at-risk, Feldman said.
Reisa Sperling of Brigham and Women’s Hospital, Boston, questioned why someone with a mutation but no biomarker evidence could be labeled as presymptomatic, while another person with biomarker evidence of AD would only be considered at risk. “Disease is in the brain, and you can’t simultaneously have the disease and be at risk for the disease,” she said.
Feldman said that as biomarker predictions become more accurate, the IWG’s definition of presymptomatic disease could expand to include certain biomarker signs. “It’s possible that neocortical tau will reach that deterministic threshold, but we’re not there yet,” Feldman said.
Why is the distinction between presymptomatic disease versus asymptomatic risk important? Feldman said that of the 400 million people worldwide estimated to have abnormal AD biomarkers, only a quarter are cognitively impaired (image above). “Diagnosing based on biomarkers could create many ‘patients in waiting’ who will never develop dementia,” he said.
Gil Rabinovici at the University of California, San Francisco, raised another concern, noting that amyloid immunotherapy is more effective at earlier disease stages. Once people are symptomatic, they have extensive pathology. “There’s the potential that by the time we diagnose the disease, the pathology will be so widespread that any intervention will be less effective,” he said in Madrid. Feldman agreed that if the secondary prevention trials now underway show treatment benefits in asymptomatic people, then the diagnostic calculus will shift. “We expect that will be a game changer,” he said. In other words, this difference between criteria may become moot once preventative treatments are available.
In the meantime, some clinicians favor the IWG approach, even among biomarker and preventive therapy enthusiasts. “Upon much consideration, we came down on [the IWG criteria] being better for our patients at this point,” Jonathan Schott of Queen Square Institute of Neurology, London, told Alzforum.—Madolyn Bowman Rogers





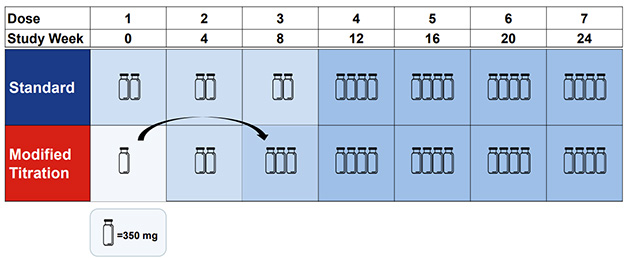























Comments
No Available Comments
Make a Comment
To make a comment you must login or register.