In October 2009, Alzheimer's researchers from around the world spent a day with caregivers and patients of families with dominantly inherited Alzheimer disease. They exchanged not only scientific news about the latest in preclinical detection of AD, but also harrowing perspectives about what it's like to live with a form of AD that strikes in generation after generation with a cruel 50 percent chance. These families are excluded from drug trials, even though they arguably hold the key to test prevention of AD. That exclusionary stance is now changing as the science and the families themselves are coming together. Read Gabrielle Strobel's seven-part update of the DIAN registry and related talks at the 7th Leonard Berg Symposium held at Washington University, St. Louis, Missouri.
St. Louis: Scientists, Families Target Preclinical Detection, Trials
On 1-2 October 2009, 214 researchers from the U.S., Europe, and Australia, as well as representatives of families with aggressive genetic forms of Alzheimer disease (AD), met at Washington University, St. Louis, Missouri, to devote two intense days of talks and discussion to their goal of presymptomatic detection of this disease. By a growing consensus, defining the “silent” phase of AD has become the field’s central challenge if it is to modernize drug testing and lay the groundwork for AD prevention trials. Presymptomatic carriers of deterministic AD mutations arguably harbor a unique potential to solve this challenge for all of AD. The St. Louis conference pulled together the latest research advances in this area. More than that, it also served as a chance for the far-flung participants in the Dominantly Inherited Alzheimer Network (DIAN) to push their ambitious project forward. In short, initial numbers on enrollment, passionate family input on counseling, community building and data privacy, and budding plans to offer a clinical trial to participating families were the main upshot of these conversations.
In St. Louis, DIAN leaders invited their external advisory committee to review and critique the study’s efforts so far. These scientists will send a detailed assessment directly to the study’s funder, the National Institute on Aging (NIA), but Thomas Bird of the University of Washington, Seattle, told ARF this: “I think the DIAN is a good idea and will provide valuable information. As I see it, the major goal is to identify the earliest signs, symptoms, and biomarker changes in persons known to be at essentially 100 percent risk for developing AD. This data will be important for understanding the biology of the disease and organizing appropriate therapeutic trials. The challenges are several: 1) recruiting and retaining an informative number of subjects, 2) choosing the “right” biomarkers, and 3) demonstrating that the information is relevant to the common forms of AD occurring in the general population.”
Presymptomatic detection in dominantly inherited AD became this year’s topic of the 7th Leonard Berg Symposium, a biennial conference at WashU that began in 1997. The symposium honors the late Leonard Berg, the founder of the WashU Alzheimer Disease Research Center. A clarinetist and saxophone player, Berg finished medical school in his native town of St. Louis at age 22 and built a life of pioneering clinical and advocacy work from there. Berg developed the widely used dementia staging instrument, Clinical Dementia Rating (CDR), which uses informant interviews and intra-individual change as important components. As early as 1979, Berg led the launch of large, decades-long natural history studies in AD and normal aging that are ongoing at WashU. Now led by John Morris of WashU, these studies have spun off longitudinal biomarker studies in the second generation of research participants and in families with autosomal-dominant AD. Berg succumbed to strokes caused by amyloid angiopathy in 2007 (for more on Berg’s life and work, see Morris and Landau, 2007).
The symposium combined a scientific update on catching presymptomatic AD in its genetic and sporadic forms (see Part 3 of this series) with discussion on whether the former models the latter, and a nuts-and-bolts meeting among the leaders of DIAN. This initiative is building an intercontinental, 10-center registry of mutation carriers and their non-carrying relatives, who are adult children of a parent with a known causative mutation in the APP or presenilin genes. It aims to pull these rare and dispersed families together into a longitudinal study that establishes the biological and cognitive course of their preclinical period with standardized data. Importantly, it tries to do so with a degree of statistical power that can support future prevention and treatment trials relevant to the families themselves and also to the much larger population of late-onset AD. The Alzforum has described the rationale and the structure of this complex undertaking before (see DIAN series); hence, this set of stories will focus on what’s new since then.
Chief among the news, DIAN is up and running. Twenty-one participants to date have completed the first round of assessments. As often happens with multicenter studies, enrollment was slow off the blocks, even as prospective participants were impatiently waiting as centers worked to obtain the necessary IRB approvals and local certifications, investigators and some family members said. Like with the Alzheimer's Disease Neuroimaging Initiative (ADNI), for example, each DIAN site needs to be certified on PIB synthesis on best practices, an imaging phantom has to be sent around to ensure standardization of the MRI measurements, etc. This has taken the better part of this year at many sites, but five sites (WashU; BWH; Brown; University of New South Wales in Sydney, Australia; and UCLA) are enrolling already and all others will begin within the month, said Morris, the principal investigator for DIAN. The goal now is to have 100 people enrolled by next July, when the DIAN leaders will gather for their forthcoming face-to-face meeting at the next ICAD conference in Honolulu, Hawaii, and 150 by end of 2010. “This number would put us on track with the original aims of the grant application,” Morris added.
The next ICAD conference also is a goalpost for perhaps the biggest new development at DIAN. At WashU last week, its leaders approved formation of a Clinical Trials Committee, which in plain English means they are putting their thinking caps on for real. Offering drug trials, either for prevention and/or treatment of early symptoms, has always been very much in the investigators’ minds, Morris said. After all, that’s what the families ultimately want and where the potential lies for late-onset AD, as well. But the investigators first needed to focus on getting DIAN going, and felt it unethical to mention future trials as part of what is primarily an observational study when it was not clear yet whether DIAN would happen or whether companies would show sufficient interest. In practice, DIAN does not have enough participants to begin a trial within the next 12 months. However, preparing for such trials requires a lot of work because challenges about design, choice of drug, and genetic privacy need to be overcome (see, e.g., Ringman et al., 2009 and 7 of this series); hence, the time to start planning is now.
Randall Bateman of WashU heads this group. “People at the steering committee meeting were very enthusiastic about starting to explore how clinical trials could be done soon,” said Bateman. At the Leonard Berg symposium, Bateman, Nick Fox of University College, London, UK, John Ringman of the University of California, Los Angeles, Stephen Salloway of Brown University in Providence, Rhode Island, and Reisa Sperling of Boston’s Brigham and Women’s Hospital discussed how best to engage drug company researchers to ensure both scientific rigor in design and execution, and independence in selecting the best drug candidate. “The hope is to have a full group, a charter, and a process in place by next July, so concrete design can go from there,” Bateman said.
During the Leonard Berg Symposium, the spouse of an eFAD patient was asked whether her adult children would want to participate in a treatment or even a prevention trial. Unsurprisingly, perhaps, the answer was: “They would try anything. Their outlook is so bleak. They worry less about side effects from an experimental treatment than about AD.”
A separate goal where DIAN is moving more slowly is expanding the network to include more sites. From the get-go, researchers in Spain, Sweden, Japan, and Italy, who have for years cared for and studied families with autosomal-dominant AD, expressed interest in joining DIAN. “We are open to expanding DIAN but are not ready yet. We first have to focus on getting all 10 sites going full steam on the existing protocol,” Morris said. Taking on more sites and more languages will add complexity and cost. One delicate question is whether sites would be added to the 10 present ones or whether a performance review might swap out underperforming sites for new ones. In St. Louis, the DIAN leaders decided to postpone the issue for one year. That said, some sites have a lot to offer the network if at least a Spanish language version of DIAN could be prepared, Morris noted. Two existing sites, Columbia University and UCLA, are working with Puerto Rican and Mexican families, respectively, and in the scientific session of the symposium, Raquel Sanchez-Valle of the Hospital Clinic Villarroel in Barcelona, Spain, reported that her team is already working with several dozen at-risk relatives from nine eFAD families (see Part 3 of this series).
DIAN itself is in large part about the families in the network. The study will be more successful if it actively integrates them into a shared sense of commitment. To this end, DIAN reserves two seats on its steering committee to family representatives. In St. Louis, an at-risk daughter and a caregiver and mother of six made their mark in two areas. First, they insisted that data sharing has to be devised with the utmost concern for their privacy. “They are steadfast on this issue,” Morris said. The Alzheimer's Disease Neuroimaging Initiative, on which DIAN is modeled, prides itself on making its data available almost instantly to all qualified researchers worldwide as soon as it enters the system. DIAN cannot do that. The investigators have to ensure that the data cannot accidentally identify families or unmask individuals within a given family. For example, study participants who chose not to know their carrier status must be unable to recognize their scan and realize they have amyloid in their brains. Therefore, the goal is to refine de-identification procedures and release that data within at most a year of its deposition in the DIAN database. “That said, we do intend to release all data, not just bits and pieces. But the guiding concerns have to be confidentiality, confidentiality, and confidentiality,” Morris quipped.
Secondly, the family representatives made a case for psychological and emotional support (see Part 2 of this series). This aggressive form of AD can divide extended families as much as it sometimes unites others. Stigma, fear, and the sheer burden of caregiving drive them into isolation. To address these needs, DIAN formed a participant liaison committee. It will work on communicating with participants and supporting them with online services. One idea is to create a forum where they can network and talk about their experiences with AD and DIAN, without the DIAN investigators being privy. “We know we ask a great deal of the study participants. We want to do things not only appropriately but also give them the necessary psychological and social support,” Morris said. Martin Rossor of University College London, UK, who has taken care of families with dominantly inherited AD for 20 years and is now seeing adult children of his original patients, will head this participant liaison group.
When asked whether support services were important, a different, unaffected spouse and mother of three adult children at risk for eFAD wrote to ARF: “Psychologic counseling, above all genetic counseling, are extremely important. The decision to find out one’s fate can be daunting, especially in the case of FAD where there is no cure. I have seen that spouses, siblings, or parents may try to influence participants to be tested or not, but ultimately it should be the decision of the possible carriers, and counseling helps them make that call. Social/emotional support from others going through the same nightmare is as important as counseling. Most families don’t know other families facing similar struggles because the genetic AD mutations are so rare. Most DIAN participants have been caregivers to their own parent or other relatives and know all too well the devastation Alzheimer’s causes to the afflicted individual and the entire family unit. I would hope for call-in support groups, but if that is not possible, then an online forum would be the next best thing.”
In other DIAN news, the scientists decided to add a new sequence to the MRI component, i.e., diffusion tensor imaging. This was not part of the initial protocol but may help the scientists understand if and when the brain’s white matter changes in the run-up to AD. This will not add imaging time or the number of sessions for participants.
Finally, DIAN has a resource allocation review panel headed by David Holtzman, through which scientists interested in studying CSF or plasma samples can request access. Holtzman, like all DIAN investigators, sites, and partnering groups, can be contacted through the Dominantly Inherited Alzheimer Network (DIAN).—Gabrielle Strobel.
This is Part 1 of a seven-part series on presymptomatic detection. See also Parts 2, 3, 4, 5, 6, and 7.
St. Louis: The Family View—What Do Study Volunteers Want From DIAN?
Scientists experiment in labs, doctors diagnose and adjust meds, company folks make drugs and run trials, and caregivers deal with the disease at home. People work, often extremely hard, in their respective corner of the Alzheimer’s world, yet they rarely come together under one roof to tell the others what it’s really like. In particular, when do researchers get to hear from study participants how deeply their families have struggled for decades and what kinds of services would make the repeated poking, measuring, and scanning of a six-year research study well worth their while?
Exactly such a conversation took place at the 7th Leonard Berg Symposium held 1-2 October 2009 at Washington University, St. Louis, Missouri (see Part 1 of this series). Unsurprisingly, perhaps, this hour stirred much more emotion than do the usual proceedings in scientific auditoriums. As two family representatives briefed the audience, both their tears and their dignity prompted spontaneous applause and standing ovations from the 214 people in attendance, most of them scientists.
The two women have seats on the steering committee of the Dominantly Inherited Alzheimer’s Network, and in this role help shape this study as it unfolds. In their talks (see below), they urged DIAN researchers to find ways to engage the participating families in broader ways. They made clear that having dominantly inherited AD reappear in generation after generation casts a wrenching pall on all family members, not just the affected relatives. Consequently, DIAN would do well to provide psychological and emotional counseling and support for people at risk for dominantly inherited AD. This could be done in person, through call-in support groups, or through protected social networking forums that build a virtual community for study participants spread across the globe. If this kind of support accompanied the study itself, one caregiver said, families would view DIAN participation as less stressful, would become invested in the effort, and more readily return for assessments. Families also need practical advice on issues such as obtaining long-term care insurance policies or persuading the Social Security agency that their loved ones qualify for disability even if they are only 45 and can still walk and talk. For their part, DIAN scientists said they would explore ways for DIAN participants to connect and speak openly about their experience without having to go through the DIAN investigators. Below are excerpts of their presentations.
Family Representative 1 (Name Withheld)
“I am showing you here a picture of my grandmother and my mother. And here’s my special auntie, mom’s oldest sister. All three were teachers. I am a teacher. All have had AD. I am at risk.
“I am 40, a wife, a mom of three young boys. I came to the U.S. on a scholarship to study special education and currently am a full-time teacher. I worked part-time in my state’s chapter of the Alzheimer’s Association, doing outreach to Latino families. Then that was becoming too much—to deal with AD all day at work and go home and deal with it personally. I needed to go back to a more normal life.
“I am mother, daughter, sister, niece, granddaughter, cousin—and the disease marks all these relationships. I work a lot on my family genealogy and it, too, is threaded through with this disease.
“I started to learn about AD from my grandmother. I did not know her well. She was in her early sixties and already very sick when I was a child. The onset in my family is in the fifties. We played school and I thought it was a game, but mom told me that granny really believed we were her students.
“I had an inkling about my mom very early on in her course. That was terrible, because I’d seen the emotional toll the disease took on my mom, who cared for grandma while also blaming her. I helped take care of grandma. One of my secrets for many years was that I was relieved when she died. I was 14 and felt so very guilty. To this day this memory makes me cry. But she was granted the gift of death. In my family the disease runs its course for some 20 years after it has fully manifested. It initially moves slowly, and that is good for us because we get to have our loved ones with us longer. But the final dreadful stages drag on for a long time.
“Alzheimer disease was stigmatized. We took care of grandma without talking about it. I found that people were hiding their sick family members. Not intentionally, but you just did not talk about it. Even in my own extended family, people have gone through it alone all the way to death. They did not know, and did not want to know, that other relatives were going through the same thing, living just a few miles away.
“In my family, people believe it is a curse visited on the family. That’s common in my culture. But even independent of culture, fear, paranoia, and confusion are all very common in families in our situation.
“We need support to deal with those feelings, particularly when we decide to participate in a research study and face our risk and fears head-on. I would welcome counseling or a support group for people like me. There is not much out there for us.
“By participating in DIAN, we are trailblazing the way into a new era of medicine. But we also need research on the psychological impact on families of autosomal-dominant Alzheimer disease. I don't know if I have the gene. No one in my family has found out. I have not because I would not do anything differently if I knew. My sister wants to know yesterday. I will want to know at some point in the future.
“What do the families participating in DIAN need? Counseling. Psychological and emotional support. Please offer it throughout the length of the study. To the researchers, I say: I am sure that if you build a good relationship with us, we will want to come back. Taking care of us in this way can prevent catastrophic outcomes.
“Around our coffee table, we are afraid of loss of insurance. We need guidance on how to navigate that issue. We are afraid of breach of privacy—please handle our data with the greatest care.
“I spend a lot of time wondering how my participation in such a study will affect my children in the future? Please take time thinking this through with us, and explaining where you can.
“From our perspective, anything and everything that makes the process more comfortable or less stressful will help the study be a success.”
Denise Heinrichs
“My husband Vince and I started our search for understanding AD in the 1960s. His mom had had it. She was diagnosed with ‘hardening of the arteries,’ the same diagnosis given to her own mom and three brothers before. Vince knew that was not it. They had died at an early age in state mental institutions or nursing homes. The relatives in his mom’s generation refused to help us. They would not talk about a mental illness. We got more help from the next generation, when several cousins died in their forties and fifties. They allowed autopsies and we found out the genetic cause.
“Then in the early 1980s, I noticed Vince’s problems. He was 39 and a college teacher. Four years later he could no longer teach. We have six children. He enrolled in an NIH study right away and got evaluated every six months. He did every test they had available at that time. At his first visit in 1983 his IQ was 136; by his last visit in 1987 it was below 50.
“He learned all he could about AD. When he could no longer drive, he rode his bike around town. It was hard for him to give up his car, but he had gotten into an accident. He read and we played Scrabble as much as we could to preserve what was still there.
“It was not easy. Our children were very aware. They were teenagers, with all the typical teenage issues, and the youngest was in kindergarten when Vince got sick. He first read to her, then she read to him when he no longer could.
“He got very angry. As with every large family, there was a lot of noise and commotion in our house, and he could not cope with that. It got hard to care for him. Stress and noise upset him. He developed seizures. In 1987, we had to place him in a nursing home. He no longer knew us and had become violent with the kids and me. That was exactly what he’d worried about early on.
“By this time, his brother already was in a nursing home with a feeding tube and was being treated for pneumonia three, four times a year. This went on many years. Vince had seen this in the years before and decided against such treatment. I made sure this would not happen to him. I obtained power of attorney and made clear that we wished no treatment other than comfort. Thankfully, he got pneumonia at some point and passed soon after surrounded by loving care and his family.
”My children are brave. They will be part of the DIAN study. I have grandchildren. My brother-in-law’s children, of course, are facing this issue, too, as are many more in Vince’s extended family.
“Alzheimer disease has consumed half of my lifetime. The toll on families is devastating, causing many additional illnesses. One problem is getting the affected on disability. This can take over a year. The wait causes immense stress and cost, and worry about losing the home or meeting day-to-day expenses. That can be overwhelming.
“I feel helplessness, sadness, fear, anger, and gratitude. I am helpless and sad for my children. I am angry because for years I have been told the cure is 10 years away, but that has not been true. I am angry as more of us get diagnosed. I am grateful for my family. I have found that family support is crucial. The parts of my extended family that support each other actively and openly are doing so much better. I am also grateful to be involved in research that is going on. We will continue to give until the answer is found. We will go anywhere and do anything until a treatment is found to stop this horrible disease. We understand it takes research participation to get there.”
Heinrichs has written a book chapter about Alzheimer’s that is available through Google books.
And here is a third voice, from a caregiver and mother of three who brought her affected husband to the symposium. When asked what DIAN could do to ensure that her children would want to enroll and keep returning for visits and invasive procedures, she said: “The single most important thing DIAN has to do to ensure return visits is to offer continued hope for the future of a world without Alzheimer’s, or at least the ability to delay the onset of the disease. The researchers and participants need to have open lines of communication and form a partnership working together for the common goal. Part of offering hope is allowing participants the first opportunity for new treatments that have been deemed safe. As an unaffected parent, I have encouraged my children to participate because I feel it is their responsibility to themselves, their families, their communities, and the world.”
Vice versa, the Leonard Berg Symposium also taught the attending families about the complexity of the science underlying dominantly inherited AD (see Parts 3, 4, 5, and 6 of this series), as well a drug company perspective on the promise and challenges of designing treatment and prevention trials for their loved ones (see Part 7 of this series).—Gabrielle Strobel.
This is Part 2 of a seven-part series on presymptomatic detection. See also Parts 1, 3, 4, 5, 6, and 7.
St. Louis: Cognition Pre-dementia—Like eFAD, Like LOAD?
At the 7th Leonard Berg Symposium held 1-2 October 2009 at Washington University in St. Louis, Missouri, scientists and patient advocates came together to exchange the latest news on detecting disease presymptomatically and to compare what’s known in rare genetic and in common forms of the disease. There was a palpable sense of excitement in the auditorium that, overall, enough lines of evidence are coming together to make preclinical detection possible in practice. Paradoxically, perhaps, this optimism arose alongside a general acknowledgment that autosomal-dominant AD is surprisingly heterogeneous, and that its status as a faithful model for the much larger population of general late-onset AD is far from cut and dried. Here are some highlights.
Ira Shoulson is a leading neuro-geneticist and clinician for Huntington disease (HD) at the University of Rochester School of Medicine and Dentistry, New York. He led the conference with a keynote on what the HD experience can teach the AD field. In HD, predictive genetic testing has been possible since 1983; hence, scientists in this community have had more time than their colleagues in AD to study the acceptance and psychological effects of such testing, as well as the characteristics of the presymptomatic period of carriers. Even so, research into preclinical HD is arguably less advanced, partly because fewer large-scale studies exist to observe how the natural history of HD unfolds. In AD, early stage diagnosis is becoming routine, at least at some academic tertiary care centers. There, clinicians try to implement biomarker-enhanced diagnoses of “prodromal” AD (Dubois et al., 2007) or similarly early stages called by a different name (WashU clinicians would call it incipient or very early AD), and the amnestic subtype of the MCI clinical categorization system appears largely to capture the same group of people. Increasingly, specialized centers will diagnose people who do not meet criteria for dementia if they have a mild memory complaint and a pathological CSF or brain scan. The rallying call in AD now is to move the diagnosis back even further, before the first symptoms appear. In contrast, the diagnosis of Huntington’s in the past 20 years has moved back only a year or two as clinicians have become more astute at recognizing early clinical signs, Shoulson said, but it is still an entirely clinical diagnosis made much later than he would wish.
In HD, people on average live 40 years of their lives at risk and 20 more years with the illness, though age of onset varies. For each person with HD, five are living at risk for the disease, but no robust biomarker signatures are in place to identify “silent” disease in these people. Scientists do know that certain abnormalities, such as cortical thinning, predate symptomatic HD, as do certain cognitive impairments, but in practice this has not translated into presymptomatic diagnosis and prevention research. To gather more powerful data on the HD preclinical phase, researchers collaborated to launch PHAROS, an observational study that to date has followed nearly 1,000 at-risk, presymptomatic people for five years. One-third of them have the extended glutamine repeat and will develop HD. “We asked people if they’d come back every nine months for evaluation. We would not share with them what we found, and we wanted their blood and put it in a database. This was very daunting, particularly securing confidentiality and privacy. It was, in part, what led to the passage of Genetic Information Nondiscrimination Act (GINA),” Shoulson told the audience (see ARF related news story on GINA).
PHAROS appears to have been worth the effort. Its initial data show that all motor and cognitive domains monitored indeed reveal abnormalities in carriers compared to their fellow non-carriers. On most tests, the differences grew over time, whereas in some tests they were present from the first assessment and then stayed constant over time. Importantly, however, on each test the carrier vs. non-carrier curves were separate throughout the duration of the study, showing that carriers already do a little more poorly in those functions that will develop overt symptoms years later. “People carrying expanded repeats did worse at baseline and worsened more over time on both motor and cognitive scores,” Shoulson said. This is instructive for the Dominantly Inherited Alzheimer Network (DIAN), which will also compare carriers to non-carriers in the decade or so before disease onset. DIAN, however, uses a different panel of assessments more heavily weighted toward imaging and CSF biomarker data. The PHAROS study stored no CSF but did store serum and urine samples; no data are available from the fluids yet. In PHAROS, as in DIAN, the hope is to use the data to identify subsets of people who are within five years prior to their diagnosis and offer targeted drug trials to them.
John Morris then summarized the rationale for DIAN and added to it the latest data in the WashU group’s ongoing studies to define exactly a detectable biological identity of those five years. Scientists have known for a decade that a sizable fraction of aging people have pathological AD—amyloid plaques and neurofibrillary tangles—without the disease’s symptoms (Price and Morris, 1999), and in the past five years, amyloid imaging has vividly confirmed this finding. Many scientists have interpreted this to mean that there likely exists a long, clinically silent stage during which AD pathogenesis operates biochemically and neurobiologically in ways that should be identified for diagnosis and early intervention. “Our efforts since then have been to characterize that stage,” Morris said.
The finding that people live with AD pathology but no symptoms also renewed a debate about whether perhaps that pathology is benign. It is absolutely not, Morris said emphatically in St. Louis. In his view, people with this pathology will develop the symptoms of AD. Exactly when that happens in a given person depends on factors such as their brain and cognitive reserve, inflammatory state, vascular health, and perhaps exercise and other environmental factors. Other speakers appeared to agree with this view (see Part 6 of this series).
New data to advance this old debate is coming on several fronts, Morris said. For one, an upcoming paper by Martha Storandt and colleagues at WashU found that, even in people who do not meet criteria for dementia, the presence of amyloid in the brain as seen by PIB PET imaging comes with a sharp decline in global composite cognitive performance (Storandt et al., 2009). For another, a WashU study led by David Johnson (now at the University of Kansas) and James Galvin reported in the October Archives of Neurology a measurable drop in cognitive performance among 444 volunteers during the transition from healthy aging to symptomatic AD (Johnson et al., 2009). People’s performance curves suddenly veered downward three years to one year before they got clinically diagnosed, even though the clinicians at WashU strive to pick up AD early. Notably, visuospatial performance caved in first, suggesting that a sole focus on episodic memory in clinical testing may miss earlier telltale signs. Episodic and working memory declined measurably in the year before diagnosis.
This result echoes studies from a growing group of other investigators who find changes in visuospatial and other domains years before a person becomes impaired enough to receive a clinical diagnosis. For example, at last July’s International Conference on Alzheimer’s Disease (ICAD) in Vienna, Jean Francois Dartigues of the University of Bordeaux, France, presented extensive and similar data from Paquid, a community-based natural history study in the Bordeaux area that has assessed 3,777 elderly people every other year for 14 years. In Vienna, Dartigues reported that people who were eventually diagnosed with incident dementia by DSM-IV criteria (i.e., years down the road from when they would get diagnosed at WashU) started declining subtly in processing speed, semantic memory, and verbal fluency some 12 years prior to formal diagnosis. Other, broader domains of memory and executive function commonly declined a few years thereafter, all of which preceded depression and the person’s own sense that something was wrong (Amieva et al., 2008). At ICAD, Dartigues detailed declines in multiple cognitive tests preceding an AD diagnosis that clearly differed from the curves of normally aging people. Both studies imply that studies of normal aging may contain a sizable fraction of people who in fact have preclinical AD, and thus perhaps overestimate the effects of truly healthy aging.
Storandt’s, Galvin’s, and Dartigues’ data comes from prospective work in either self-selected or community-based elderly people. What about dominantly inherited AD? At the Leonard Berg Symposium, Martin Rossor of University College London, UK, noted that from the very beginning of research with carriers of APP and presenilin mutations, changes in verbal, visual, and spatial memory were found to precede a broader deterioration that signaled the onset of clinical AD (e.g., Newman et al., 1994). Francisco Lopera is a neurologist at Antioquia University School of Medicine, Medellín, Colombia, who works with the world’s largest known pedigree carrying a presenilin mutation and attended the Leonard Berg Symposium. His research has pointed to verbal and semantic memory as early indicators, as well. And John Ringman of the University of California, Los Angeles, found stark differences in both the trail making test (which taxes visuospatial and executive speed) and verbal memory tests when he compared 30 mutation carriers and 21 non-carriers from 10 Mexican families with presenilin mutations. These differences between non-carriers and carriers arose with age; they were not evident when carriers were still 20 years away from their family’s expected age of onset but were about five years prior (Ringman et al., 2005). Since then, a newer study of 42 Mexican American people at risk of a pathogenic presenilin mutation has begun, and many of these subjects are opting to participate in DIAN, Ringman said in St. Louis. While these tests broadly point to the same functional domains, getting scientists to agree which tests to use will be one of the bigger challenges of any joint prevention initiative, several speakers at the Berg Symposium acknowledged. To name but a few, cued semantic recall tests and attention tests measuring intrusion errors are also increasingly cropping up as sensitive indicators very early on, and cognitive psychologists are known for fiercely debating the pros and cons of their favorite tests.
Based on this and other data, it seems that what scientists have found out about the preclinical cognitive decline in autosomal-dominant familial AD largely jibes with findings from late-onset AD, Rossor and other speakers said. “Cognitive decline in early familial AD is similar to that seen in LOAD,” said Ringman. This, in turn, holds out hope that biomarker data from the DIAN network may likewise be generalizable to the common forms of AD. A predictive biomarker signature is indispensable because longitudinal studies aren’t an option for the diagnosing physician, who ideally needs to get the job done in a single visit.—Gabrielle Strobel.
This is Part 3 of a seven-part series on presymptomatic detection. See also Parts 1, 2, 4, 5, 6, and 7.
St. Louis: Biomarkers Pre-dementia—Like eFAD, Like LOAD?
It is a sign of progress in Alzheimer disease research that longitudinal observational studies are beginning to converge on when and how a person’s cognition shows the first subtle signs of trouble on the way to dementia (see Part 3 of this series). That’s dandy for research, but clinicians urgently need robust tools to diagnose presymptomatic dementia in one clinic visit. Part of the toolkit for that will come from biomarkers, and at the 7th Leonard Berg Symposium, held 1-2 October 2009 at Washington University, St. Louis, scientists shared some of the latest news in this burgeoning area of study. As throughout this conference, talks toggled between what’s known in LOAD and eFAD, comparing all the while how well knowledge on these forms of AD matches up.
John Morris of WashU started the topic with an update on his center’s Antecedent Biomarker Study, which has been seeking to find a predictive combination of biomarkers in cognitively normal adult children of a parent with AD since 2005. (These are not families with dominantly inherited AD.) Dispensing with cautious qualifiers, Morris summed up the bottom line of this work: “We can detect preclinical AD in cognitively normal older adults.” How long before dementia? About four years. And this is how it works, claimed Morris: When people have reduced CSF Aβ42, elevated CSF tau/phospho-tau, and amyloid in their brains, they will subsequently develop dementia. Their Aβ42 drops first, brain amyloid shows up soon after, and tau starts rising just prior to symptoms (for comparison, see Johnson diagram in Part 6). Almost all people whose CSF Aβ42 is abnormally low also have amyloid in their brains.
This website has covered many individual studies on the way to this conclusion (e.g., Skoog et al., 2003; Sunderland, 2003; Fagan et al., 2006; Fagan et al., 2007; Li et al., 2007; Shaw et al., 2009), as well as the broader literature showing that damage to the brain is extensive by the time a person is diagnosed. Hence, this story will focus on the latest data presented in St. Louis. For example, Morris reported that the amyloid in people’s brains is driven by the two leading risk factors for late-onset AD—age and ApoE. In a PIB PET series of 241 cognitively normal people age 45 to 89, brain amyloid started showing up around age 55 and became more and more frequent in older folks (Morris et al., 2009). People with ApoE4 were highly overrepresented relative to their allele frequency among the PIB-positive group, whereas almost no one with ApoE2 had brain amyloid even up into the highest ages. In the sixties, seventies, and eighties, the percentages of people with ApoE4 being positive for PIB were 37, 53, and 75, respectively; for people without ApoE4, these numbers were 8, 16, and 16 percent by comparison. “This suggests that expression of the E4 phenotype is very strongly associated with presence of cerebral Aβ deposits as people age,” Morris said. In essence, this makes brain amyloid a phenotype of sorts of ApoE4 (see also Reiman et al., 2009). And an upcoming paper in Archives of Neurology reports that, among a group of 159 cognitively normal research volunteers, having amyloid in the brain predicts that the person would develop AD symptoms when followed for up to 5.5 years (mean of 2.4 years; Morris et al., 2009).
Besides ApoE and age, scientists are currently looking to relate the two linked biomarkers of abnormal CSF/brain amyloid to additional known risk factors and markers of AD. The hope is that a comprehensive picture might emerge of how multiple parts of the biology fit together in the preclinical phase. One recent step in this effort was a paper showing that in cognitively normal people, the abnormal CSF signature is statistically linked with brain atrophy; at later stages, when dementia sets in, tau drives the CSF-atrophy relationship as it continues to rise while the brain continues to shrink with progressing illness (Fagan et al., 2009). More than just whole brain atrophy, brain amyloid in cognitively normal older adults is associated with thinning of the cortex in regions known to be vulnerable to AD pathology (Dickerson et al., 2009). This suggests that a preclinical CSF signature is beginning to match up with an imaging signature composed of amyloid and cortical thinning.
In his talk, David Holtzman of WashU noted that a new Dutch/MGH study reporting reduced CSF Aβ40 concentrations in cerebral amyloid angiopathy (CAA) provided yet another independent confirmation for the general idea that as amyloid deposits in the brain, in this case on blood vessels, it becomes trapped and Aβ concentrations in the CSF drop. CAA is a common cause of strokes (Verbeek et al., 2009).
An upcoming paper from the WashU group further tightens the connection between brain amyloid and CSF Aβ by analyzing CSF Aβ versus PIB and age in 189 cognitively normal people, Holtzman said (Fagan et al., in press). In this series, everyone whose brain binds PIB also has low CSF Aβ42, but the opposite is not true. Some people, especially the youngest participants between 45 and 55 years of age, already have low CSF Aβ42 but no PIB. The scientists interpret this to mean that brain amyloid deposition begins in a conformation that may initially be invisible to PIB. The subsequent drop in CSF Aβ42 would consequently be the earliest detectable biomarker at present. When the brain amyloid later becomes fibrillar, it binds PIB. “This is our impression so far, but we do not have proof yet,” said Holtzman. “We have to follow these cohorts longer to see in which order these markers come up.”
The same paper also contains more data connecting the CSF combination of high tau/low Aβ42 with brain amyloid, in essence predicting that this CSF signature reflects ongoing neurodegeneration and will predict onset of symptoms in the next three to five years. Building on a smaller previous study, this finding extends into cognitively normal people in a widely cited study from Kaj Blennow’s group three years ago, in which virtually everyone with this CSF signature among a large cohort of MCI patients converted to AD within five years (Hansson et al., 2006). In this study and an independent recent one, these biomarkers predicted not only whether people would develop AD, but also how fast their cognitive decline would progress (Snider et al., 2009).
Now, for the first time, a pharma company, Bristol-Myers Squibb, targets people at this pre-dementia stage for a drug trial. The new twist, compared to previous MCI trials (which all failed), is that not an MCI diagnosis but a low CSF Aβ42 level plus a subjective memory complaint determine whether a person can enter the trial. CSF Aβ/tau, and brain atrophy are the outcome measures listed just below safety. If the distinction between CSF Aβ42 and CSF tau changes holds up in larger studies, i.e., if the former truly precedes the latter by two years or so, then future trials could push back to treating asymptomatic people by screening for low CSF Aβ42 and enrolling people just at the point when their tau is beginning to nudge up but before they have symptoms. CSF tau could then conceivably become an outcome measure to see if the drug is effective. That, then, would constitute a prevention trial, and it may soon come within reach, said Reisa Sperling of Brigham and Women’s Hospital.
These data come from research with volunteers who have, or may develop, the common forms of AD. Establishing the order of antecedent biomarkers in dominantly inherited (aka autosomal-dominant or early onset familial) AD with sufficient statistical power to support drug trials is part of what the Dominantly Inherited Alzheimer Network (DIAN) is aiming to accomplish. Small biomarker studies with individual families have already begun to pave the way. For example, researchers led by Dan Pollen of University of Massachusetts Medical Center in Worcester, who described the first reported presenilin 1 family, reported CSF Aβ42 decreases in six presymptomatic mutation carriers (Moonis et al., 2005). Last year, John Ringman of UCLA reported the same thing, plus that CSF tau was increased. In this study, CSF isoprostanes were up, too, as was plasma Aβ (Ringman et al., 2008).
At the Leonard Berg Symposium, Raquel Sanchez-Valle of the Hospital Clinic in Barcelona, Spain, reported new CSF and plasma data on 14 relatives from four different families with presenilin mutations. Of the eight participants who carried the AD mutation, half were symptomatic, half not yet. These Spanish investigators offer genetic counseling, testing, and observational research to families with genetic neurodegenerative diseases including eFAD (see eFAD studies). In St. Louis, Sanchez-Valle presented the first cross-sectional data of what is to become a longitudinal study of these volunteers. Using Innogenetics’ Innotest for CSF, and a cutoff value of 495 pg/ml (as per van der Vlies et al., 2009), CSF Aβ42 levels were normal, i.e., high, in those presymptomatic carriers who were still more than a decade away from their family’s mean age at onset, and low in even mildly symptomatic carriers, Sanchez-Valle reported. CSF tau was elevated only by the time carriers became clearly symptomatic, and it then correlated strongly with a person’s clinical dementia rating or MMSE. This Spanish group found no differences in amyloid plasma between carriers and non-carriers in this initial study.
“Overall, this indicates that the same kind of changes are occurring in dominantly inherited AD as in late-onset AD,” Holtzman said, but cautioned that these studies are all very small.
Alzheimer's Disease Neuroimaging Initiative (ADNI), nor the Adult Children Study and other cohorts should restrict their analyses to the usual suspects Aβ and tau. A multitude of other markers are coming out of proteomics analyses of CSF and plasma, and some bear close watching. For example, Eric Portelius, working with Henrik Zetterberg and Kaj Blennow at University of Gothenberg in Sweden, has developed combined immunoprecipitation/MALDI-TOF mass spec protocols to explore the proteomic diversity of Aβ and APP species in CSF of LOAD and FAD. The Swedish scientists have found some 20 different Aβ species in CSF; Aβ42 was one of the least abundant ones. It is important to disease because it is hydrophobic and aggregates readily, but as biomarkers, other species may be easier to use and more informative. Variability among centers, particularly in Aβ42 measurements, has been dogging the field for some time (Verwey et al., 2009; Mattsson et al., 2009). At ICAD in Vienna, Zetterberg included in his plenary lecture unpublished data suggesting that in the CSF of some familial AD cases, Aβ37, 38, and 39 were all particularly low, whereas an Aβ1-16 fragment was abnormally high. This pattern differed starkly between PS-mutant AD and sporadic AD, though both forms had similar, and expected, findings on Aβ40 and 42. Aβ1-16 popped out of that work as a novel biomarker candidate for both sporadic and familial AD. “DIAN should look at these other species, too,” Holtzman said.
Also at ICAD, Zetterberg presented his group’s detection in CSF of AD patients of a set of truncated Aβ forms, as well as a set of APP fragments, both of which point to the existence of a new, yet-to-be-defined cleavage sequence of APP (see also Portelius et al., 2009; Portelius et al., 2009; Portelius et al., 2009). Beyond APP and Aβ species, a wealth of potential markers are being discovered. To quote but one example from the Leonard Berg Symposium, the poster session featured a study by Rawan Tarawneh and colleagues at WashU on the neuronal injury marker VILIP-1, an intracellular calcium-sensor that tracked with dementia severity in a small study of nine AD patients and 15 controls.—Gabrielle Strobel.
This is Part 4 of a seven-part series on presymptomatic detection. See also Parts 1, 2, 3, 5, 6, and 7.
Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka SK, van der Flier WM, Blankenstein MA, Ewers M, Rich K, Kaiser E, Verbeek M, Tsolaki M, Mulugeta E, Rosén E, Aarsland D, Visser PJ, Schröder J, Marcusson J, de Leon M, Hampel H, Scheltens P, Pirttilä T, Wallin A, Jönhagen ME, Minthon L, Winblad B, Blennow K.
CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment.
JAMA. 2009 Jul 22;302(4):385-93.
PubMed.
St. Louis: Is Rare Familial Alzheimer’s a Model for the Millions?
One question that pervaded the 7th Leonard Berg Symposium held 1-2 October 2009 at Washington University in St. Louis, Missouri, is whether dominantly inherited Alzheimer disease really is the same disease as the common late-onset forms that afflict some 35 million people around the globe, according to a recent report (ARF related news story). “Can we generalize from FAD to all AD?” asked Martin Rossor of University College, London.
The question is important in part because the interest drug developers take in testing prevention in families with strong AD genetics hinges on their confidence that success in the few will translate into success in the many. In private, some industry leaders express doubt about that point, and some companies exclude eFAD families from their trials for this reason (see eFAD Research essay). That eFAD indeed models late-onset AD (LOAD) to a great extent, if not in all aspects, is a premise of the Dominantly Inherited Alzheimer Network (DIAN), and the comparison of DIAN and the Alzheimer's Disease Neuroimaging Initiative (ADNI) data may settle the issue definitively. In the meantime, scientists at the Symposium discussed what clues they have so far. See below for talks taking stock of LOAD/eFAD similarities and differences in the clinic, pathology, biochemistry, genetics, and imaging.
First, a word about nomenclature. Autosomal-dominant Alzheimer disease is the form in which a child of an affected parent faces 50-50 odds of inheriting a pathogenic mutation in either the APP or the presenilin 1 or 2 genes. It is variously referred to as “familial” or “FAD,” “early onset” or “EOAD,” “early onset familial” or “eFAD” (see ARF eFAD section). Strictly speaking, none of the terms is interchangeable. Familial AD often is merely a clustering, not a Mendelian autosomal-dominant inheritance pattern; early onset AD is often sporadic in the sense that neither affected relatives, much less a gene mutation, is known. The term used in the Alzforum section on this set of aggressive forms of AD—eFAD—does not quite catch all people it means to include, either, because recently, autosomal-dominant pathogenic presenilin mutations were spotted in some rare families who get this form of AD in their seventies, i.e., with a late onset (Kauwe et al., 2007; Brickell et al., 2007). The Washington University researchers sidestepped this confusing nameology by using yet another term: “dominantly inherited Alzheimer disease.” DIAD conveniently stands opposite LOAD and echoes DIAN, making DIAN the network for the DIAD crowd.
At the Leonard Berg Symposium, speakers stressed that DIAD/eFAD overall appears to model LOAD quite closely. However, it’s no carbon copy, and for the sake of discussion, the differences got center stage for a session. DIAD is heterogeneous. The way people decline can vary a bit from person to person, said Rossor, whose center follows members of 10 families with APP mutations and 34 families with presenilin 1 mutations. Certain clinical differences do track with genetic ones. For example, people who get DIAD because their APP gene is duplicated frequently have seizures, brain hemorrhages, and white matter changes. In these extremely rare families, some affected relatives start having seizures when they are teenagers, and depression and then dementia follow within a decade thereafter.
With presenilin, some 175 different mutations are published, but clinicians have no comprehensive description of how their preclinical period unfolds. DIAN is aiming to accomplish that and in the process may match up genotypes to phenotypes. Based on what clinicians observe, it is already clear that some presenilin cases come with added signs that are atypical for AD. For example, some patients have a paralyzing weakness of the legs called spastic paraparesis, and stumbling and falls can be among the first signs mutation carriers report. One new Bulgarian family is described in the literature this month (Dintchov et al., 2009).
Corresponding to this clinical phenotype, Bill Klunk of the University of Pittsburgh Medical School, Pennsylvania, in St. Louis showed slides of DIAD/eFAD patients with spastic paraparesis who have heavy amyloid deposition in their cerebellum, an area typically spared early on in LOAD. Pathologically, a particular kind of plaque called cotton wool plaque—shaped a bit like a fuzzy ring with a hollow in the middle—appears to match up with this symptom (Dumanchin et al., 2006; Karlstrom et al., 2008), but again, there is no definitive link between particular mutations, a particular pathology, and a characteristic clinical phenotype. Myoclonus (a form of muscle twitching) and rigidity also can be part of presenilin-mutant AD, as is Lewy body pathology (Leverenz et al., 2006). Not all mutation carriers in one family will develop these additional symptoms. On the other hand, people with DIAD lack certain deficits that are sometimes seen in sporadic AD. An example Rossor gave was impairment of visual processing and is sometimes referred to as posterior cortical atrophy, or the posterior variant of AD.
In terms of pathology, DIAD can be heterogeneous as well, said Bernardino Ghetti of Indiana University Medical School in Indianapolis. Diffuse plaques, neuritic plaques, and cotton wool plaques are the major types, and vascular deposits can be extensive in some families. Other rare families have abundant pathology in their cerebellum coupled with concomitant ataxic symptoms of poor movement coordination, and yet other presenilin mutations give rise to mixed dementia/parkinsonism reflected by cotton wool plaques in both AD- and PD-vulnerable regions. Some of the most aggressive forms of DIAN, where patients die in their thirties, show large amorphous cotton wool plaques without much neuritic structure. The vasculature in these cases is also severely damaged, with large plaques breaking through vessel walls in many places, Ghetti said. Intriguingly, Ghetti noted that these cotton wool plaques contain little Aβ1-42 but a lot of N-terminally truncated forms of Aβ, either with or without pyroglutamated residues on positions glu-3 and glu-11 (Miravalle et al., 2005).
Biochemically, the heterogeneity of the 175 published presenilin 1 mutations has triggered much debate among molecular biologists about how the mutations cause AD (see ARF Davies/De Strooper Discussion; Shen/Kelleher Discussion). At this point of the debate, a frequently heard view holds that individual differences among mutations tend to have one feature in common. That is, they disturb the function of the γ-secretase enzyme complex in such a way that the enzyme generates a pathologically altered distribution of Aβ peptide variants, Bart de Strooper of the VIB Institute in Leuven, Belgium, said in St Louis. In short, the mutant enzyme makes more of Aβ long forms and fewer of its short forms (see also Part 4 of this series). The Familial Adult Children Study (FACS) aims to capture the dynamics of this process by measuring in real time the production and metabolism of Aβ in the CSF of people with DIAD. In St. Louis, FACS leader Randy Bateman noted that 26 of the planned 36 participants in this study have enrolled as of last August, and that the last nine of those have also completed all DIAN procedures.
Several speakers hinted that the most aggressive PS mutations might even express themselves developmentally. Ghetti said that families with myoclonus and seizures have ectopic neurons in the white matter, i.e., neurons that presumably migrated to the wrong place in utero (Takao et al., 2001); John Ringman of the University of California, Los Angeles, noted a trend toward less education among PS1 mutation carriers in Mexican families he studies.
“Overall, I think FAD is actually more heterogeneous than sporadic AD,” Rossor summed up. This may be counterintuitive to conventional wisdom, which loosely views LOAD as resulting from many different age-related insults and eFAD as being due to mutation of amyloid-generating genes. On the other hand, a focus on variations and extremely rare cases runs the risk of losing the forest for the trees, Bateman wrote. “In our clinical experience, the majority of DIAD families do not have seizures, myoclonus, or other “atypical” features, all of which are sometimes seen in LOAD, as well,” Bateman wrote. “We scientists love to look for differences, but the main point we agree on is that DIAD appears highly similar to LOAD overall.”
Perhaps the most significant aspect of DIAD heterogeneity is the age span at which carriers begin to get overtly ill. Presenilin mutations can become apparent from the teenage years to the seventies. Those are the extreme ends, however; most families have a 10- to 15-year spread around a mean age of onset in the forties or fifties. Besides presenting uncertainty for the carrier and practical challenges for future prevention trials, this broad range harbors some scientific promise, too, Alison Goate of Washington University said at the Leonard Berg Symposium: “There are other things besides the presenilin mutation going on that determine age of onset. They could be genetic or environmental.” If these things could be understood and exploited therapeutically, even a modest five-year delay in the age of onset (of LOAD in this case) would be a boon to public health.
Some clues on what these factors might be have already come out of research on LOAD. Genetically, the ApoE gene plays the single biggest role, with each E4 risk allele bringing down age at onset by some five years, Goate said. ApoE interacts strongly with head injury in that ApoE4 carriers are more likely to develop dementia after they sustain brain trauma. For example, boxers with dementia pugilistica tend to have E4 alleles (Jordan et al., 1997; ARF Discussion). Moreover, genetics research has generated a long list of genes that play smaller but possibly important roles in LOAD. Whether any of these genes influence age at onset in DIAD is a question DIAN could address, for example, by looking for top AlzGene risk alleles in the participating families as well. LOAD genetics just got a boost from the discovery of three new genes this past summer (see ARF related news story), but they have not been checked in DIAD/eFAD yet. How about ApoE in DIAD/eFAD? Even this oldest LOAD risk gene has not been systematically studied in eFAD families; however, it is known that in large Colombian kindreds with the presenilin 1 E280A mutation, carriers who have E4 become symptomatic at a younger age than E3 carriers, and E2 carriers stay well some years longer still. “Even in this early onset FAD kindred, E4 influences when a carrier will get sick,” Goate said.
In terms of environmental factors, little is known in DIAD/eFAD. Research of the same Colombian pedigrees suggested, surprisingly, that people with a higher level of education were diagnosed at younger ages than less educated carriers. This was unexpected because education is thought to be protective by affording some brain reserve. In this study, the result might simply mean that more educated people were being picked up as having a problem earlier because they were performing more demanding jobs (Mejia et al., 2003). Unlike LOAD, which tends to show up in retirement, eFAD typically gets noticed first at work.
Brain imaging has advanced immensely, and numerous lines of evidence, from ADNI and elsewhere, are gradually bringing into focus a view of preclinical AD for both LOAD and DIAD/eFAD (for an update, see Part 6 of this series). Imaging has produced its share of puzzling moments, however, where it appeared to highlight the heterogeneity of eFAD. For example, in 2007, when excitement over amyloid imaging began to spread worldwide among AD research groups, Klunk, who co-developed Pittsburgh compound B (PIB) with his friend and colleague Chet Mathis, published the first PIB images of presymptomatic carriers of the C410Y and the A426P presenilin mutations. Their PIB retention began building up with an extraordinarily intense signal in the striatum, an area hit hard in Parkinson and Huntington diseases, but usually spared in AD (Klunk et al., 2007). The typical AD regions lit up, too, but much more weakly. This finding seemed at odds with the clinical and pathological picture of AD, and it prompted questions about whether eFAD truly models LOAD. “This unusual distribution was so surprising, I first accused Chet of accidentally giving me a dopaminergic agent, not PIB,” Klunk recalled in St. Louis. But PIB it was. Since then, the London researchers have observed a similar striatum-first PIB pattern in members of some of the families they follow, Rossor said in St. Louis.
In the meantime, Klunk’s group has obtained repeat scans from those and other DIAD/eFAD research volunteers. He offered this update: Over the course of four years, the PIB pattern spread out from the striatum to a typical AD pattern with increasing binding in cortical regions, whereas the original striatal binding diminished over this period of time. The pattern of PIB retention varied somewhat by mutation. Overall, Klunk said, it looks like in eFAD, amyloid deposition starts in the striatum as early as 10 years before symptoms, peaks before or around the time symptoms appear, and then spontaneously decreases. Neocortical amyloid appears later but then stays. In LOAD, amyloid deposition begins in the neocortex (in the precuneus and anterior cingulate subareas), later spreads to include the striatum, and does not decrease.
“We also see the striatal pattern of eFAD in Down’s, the only other Aβ overproduction syndrome,” Klunk said. “We think it is related to that but cannot explain it.” In the discussion, scientists noted that PIB may not see cotton wool plaques and diffuse plaques. It also does not bind non-fibrillar forms of Aβ, all of which may develop with a slightly different time course and distribution in eFAD versus LOAD.—Gabrielle Strobel.
This is Part 5 of a seven-part series on presymptomatic detection. See also Parts 1, 2, 3, 4, 6, and 7.
St. Louis: Imaging Preclinical AD—Can You See it Coming in the Brain?
At the 7th Leonard Berg Symposium, brain imaging researchers of various stripes—some MRI, some FDG PET, some amyloid imaging aficionados—took stock of what brain imaging tells the field about the preclinical period of Alzheimer disease. From their vantage point, a picture is slowly coming into focus whereby a number of different imaging techniques together predict that a person is on the way to developing Alzheimer’s symptoms. For some speakers, the evidence is strong enough to start thinking seriously about how to use images (in conjunction with CSF and cognition) to test prevention of AD in people who are at high risk, either by virtue of having inherited the ApoE4 risk allele, or a dominant mutation.
Nick Fox of King’s College, London, UK, reminded the audience that of the imaging modalities currently used, magnetic resonance imaging (MRI) is one that can yield perhaps the most exquisite view of the brain with a resolution of 1 mm. This can be had with a 10-minute scan session that is robust and practical for the imaging technician and easy to tolerate for the subject, certainly for presymptomatic people and even for many patients. This method is particularly suited for tracking change, for example, shrinking of brain areas, over time, and to measure progression. “We are all interested in drugs that slow or stop progression,” Fox said.
Some prior imaging studies on presymptomatic eFAD mutation carriers have been done, all very small (Ridha et al., 2006; Godbolt et al., 2006; Ringman et al., 2007; Ginestroni et al., 2009). Early data from Fox’s group suggest that, after diagnosis, people with eFAD lose volume rapidly. Fox showed images of one person’s hippocampus shrinking dramatically within only two years of diagnosis. Many years prior to diagnosis atrophy rates are slow, but five to two years prior, they noticeably pick up to as much as 3 percent per year in particularly vulnerable areas. Rates come in plural form because they differ by brain area; for example, the entorhinal cortex shrinks particularly fast just before diagnosis. “Serial imaging gives us fantastic insight into structural change prior to diagnosis. The excitement of DIAN is that we can pull more people together to expand on these findings with better power,” Fox said.
Technically, the large range of normal variation in brain size between one person and the next makes it important to focus on rates of change in given areas, not absolute measures, and also to focus on the area that changes between two well-registered images (aka the boundary shift interval), without including the volume of the entire brain, Fox noted.
MRI is one of the most established ways of brain imaging. In the past five years, it got increasingly prominent company from amyloid positron emission tomography (PET), which visualizes in fiery color and in living people pathology that previously could only be seen after a patient had died. Research groups around the globe quickly adopted the amyloid label Pittsburgh Compound B (PIB) or similar compounds for study, and as people began to drill deeper, the story became more complicated. Questions arose about what it means that a large fraction of cognitively healthy elderly have amyloid in their brains, about exactly which forms of amyloid PIB binds to, and how being PIB positive related to other liabilities ascribed to the preclinical stage of AD. At the Leonard Berg Symposium, Keith Johnson of Massachusetts General and Brigham and Women’s Hospitals in Boston addressed these questions with the data available to date.
Multiple studies show that, by and large, everyone with AD has high PIB retention in their brains, Johnson said. There are exceptions relating to the maturity of plaques and perhaps “strains” of Aβ (e.g., Rosen et al., 2009). “These are interesting and important, but they are exceptions,” Johnson stressed. At the early symptomatic MCI stage, people either have a control pattern or an AD-like pattern, not an in-between pattern. The so-called PIB-positive (PIB+) controls, that is, people who are clinically normal but have amyloid in their brains, tend to have it in a stereotyped AD-like distribution with a weaker intense signal. Around the time of diagnosis, PIB binding tends to become saturated and stay relatively stable through the course of clinical disease.
One burning question on the minds of people who study preclinical detection in different populations and with different methods is how the data might all fit together. Johnson offered this hypothesis:
By that scenario, the long road to disease would begin with amyloid-β pathology. Age and genetics (i.e., ApoE genotype) influence a person’s propensity to deposit amyloid, and both CSF Aβ and PIB PET can detect it. This would lead to synaptic dysfunction, which impairs cognition directly, and also indirectly over time as the neurons bearing malfunctioning synapses slowly die. It is widely accepted that brain and cognitive reserve influence how long a person can withstand the damaging effects of the encroaching pathology. Bill Klunk, of the University of Pittsburgh Medical School in Pennsylvania, noted that vascular disease is another important environmental factor that acts at this level. The PIB+ clinically normal people currently draw intense interest among researchers. Are they the ones who are on this path to AD? Imaging methods from functional MRI to FDG glucose PET and volumetric MRI exist to visualize and quantify these stages. The standard clinical tests used routinely in diagnosis pick up the latest stage on this flow chart, but increasingly, new findings are converging around more sensitive cognitive measures to pick up earlier, subtler changes (see Part 3 and below).
One way of probing whether this suggested signature of AD holds true is to test whether amyloid deposition in clinically and cognitively normal people relates to the other components in this diagram. “We do this by looking at other measures of AD in PIB+ controls,” Johnson said. Here are four examples of doing so that Johnson presented in his talk:
Cortical thickness: In MGH studies of cognitively normal PIB+ people, increasing amyloid is associated with a thinning cortex in the precuneus, anterior and posterior cingulate, and some temporo-parietal and medial frontal cortical areas. These areas have come to represent a network that represents functional connection. “These areas talk to each other,” Johnson said.
Hippocampal volume: In the MGH/BWH studies, this measure does not seem to relate to amyloid deposition in the cognitively normal volunteers but does by mildly impaired stages of CDR 0.5 and 1. “This suggests to us that the cortex thins first and the hippocampus a bit later,” Johnson said.
Brain connectivity: A recent study reported that the more highly linked an area is—in other words, if it is a hub of intense connectivity—the more amyloid it tends to have (Buckner et al., 2009). This month, the researchers reported an amyloid-related disruption of intrinsic connectivity among asymptomatic elderly (Hedden et al., 2009). “This suggests a disconnection where one area is unable to talk with other, previously connected areas anymore,” Johnson said. In July, the same researchers reported that using fMRI, they found that brain amyloid in non-demented people mapped to their inability to deactivate the default network when focusing on a task (Sperling et al., 2009).
Cognitive function: It is becoming increasingly clear that amyloid deposition is tied to cognitive impairment. IQ and education are important here because people high on those measures are able to tolerate amyloid pathology longer. People with low IQ/education scores show a much stronger link between amyloid in their precuneus (a particularly early area) and how they perform on neuropsychological tests (Rentz et al., Annals of Neurology, in press). And preliminary analyses of longitudinal data showed a detectable decline in a delayed word recall test. People declined along with increasing precuneus PIB. These are subtle effects on difficult tests, Johnson noted; the first difference the MGH/BWH studies see in those tests is that people with PIB in precuneus lose the practice effect, i.e., they don't improve anymore with repeated testing. “We really don’t understand in biological terms what cognitive reserve is and how it preserves function,” Johnson noted.
The Society for Neuroscience conference, held 17-21 October 2009 in Chicago, featured even newer data along these lines. To quote but one example, Deepti Putcha, working with Reisa Sperling at Brigham and Women’s Hospital in Boston, reported that cognitively normal older people who performed poorly on a challenging memory test (RVALT) had hyperactivation of their hippocampus along with cortical thinning in AD-vulnerable areas of the medial temporal lobe. These data are fresh out of the scanner, and concomitant amyloid and connectivity imaging data on this new aging cohort still needs to be analyzed, Putcha said.
These and other emerging clues on how multiple preclinical markers link up together in the same people will hopefully bring into focus a predictive signature over the course of the next five years, as longitudinal studies under way at several institutions progress. But even now, the field has advanced far enough to start planning prevention research, argued Eric Reiman of the Banner Alzheimer's Institute in Phoenix, Arizona. Evaluating promising presymptomatic treatments in the general population takes too long and is too costly, but doing so in high-risk groups such as ApoE4 carriers and presymptomatic carriers of dominantly inherited Alzheimer disease (DIAD)/eFAD mutations has come entirely within reach, Reiman said. Adding to the other speakers’ presentations a brief summary of his group’s research, Reiman noted that he has followed ApoE- genotyped volunteers—that is, people at three defined levels of risk—with brain imaging for well over a decade. People with the ApoE4 allele have decreased glucose utilization and subtle metabolic changes already as young adults, predating even the earliest biochemical marker to date, i.e., CSF Aβ. These FDG PET abnormalities do not progress continuously from early adulthood, but they flag early on where the AD changes will develop later, Reiman said (Reiman et al., 2001; Caselli et al., 2004; Reiman et al., 2005; Reiman et al., 2009).
The wealth of imaging and biomarker data the field has accumulated to date provides a sufficient foundation for proof-of-concept trials of biomarker outcomes, not clinical outcomes, with several hundred subjects, Reiman said. He spoke forcefully for starting an era of prevention research. This will not be a quick achievement, but it is time to begin gathering drug/biomarker data that can open a new regulatory path to approval. Such trials would also show research volunteers who have allowed scientists to poke, question, and scan them for decades that the scientists, in turn, will use all that data toward something tangible that truly matters to the volunteers and their families.—Gabrielle Strobel.
This is Part 6 of a seven-part series on presymptomatic detection. See also Parts 1, 2, 3, 4, 5, and 7.
St. Louis: An eFAD Prevention Trial—One Man’s View
The Dominantly Inherited Alzheimer’s Network (DIAN) was founded with the hope that its participating families could be offered a prevention trial and/or treatment trial before too long. Patients with eFAD are routinely excluded from drug trials because they tend to be younger than most trials allow and because many trials explicitly bar patients with APP or presenilin mutations. There is a bitter irony in denying drug trials to the very families whose research participation has helped open up the genetics and ensuing molecular biology of Alzheimer disease research, built countless careers in the process, and led to the development of those very drugs. This situation is finally beginning to change. Even just three years ago, leading company scientists still overwhelmingly focused on the reasons—regulatory, scientific, practical, ethical—why such trials weren’t happening (see eFAD Research section). Since then, the buzz has become noticeably more positive. Researchers across the field now openly recognize the promise of testing prevention in high-risk groups such as eFAD, and the language has changed from ‘Would be nice but can’t do it’ to ‘Want to do it. Planning how.’
For its part, DIAN was formed without industry researchers to ensure it could be independent in choosing the best candidate drugs for this small population, and its newly formed exploratory drug trial committee (see Part 1 of this series) has no pharma representatives, either. But informal consultations are going on. At the 7th Leonard Berg Symposium, Dale Schenk of Elan Pharmaceuticals in South San Francisco addressed the audience of scientists, caregivers, and early stage patients. Schenk has for years expressed an interest in finding ways to do such trials, and here, summarized, are his remarks:
“To the family members who came, I say it’s all about you. We have worked on making an anti-amyloid drug for 20 years and we hope to be right at least once, to make at least one good drug.
“Yesterday I went to the Lewis and Clark exhibit here in town and felt some kinship in what we are trying to do. They headed out on a journey not knowing what they’d find; they got hit by arrows but also had moments of stunning beauty along the way. I can assure you we are hoping for those moments, too.
“I will present to you a few considerations about potential treatment and drug trials in dominantly inherited Alzheimer disease (DIAD). They are not answers, but rather questions as we think about agents that we might consider useful in delivering to FAD patients.
“To date, most of what we do know in the clinical arena is about sporadic AD. Almost all trial paradigms are in mild to moderate AD. The entry criteria is clinical AD; we use dual endpoints—a cognitive and a functional one. And by definition, studies are clinically-based. Imaging and biomarkers are used at best as secondary endpoints, not primary ones. We need to move beyond that.
“We all want to go to prevention. How to do that? We go earlier and earlier. The next step is mild AD and those MCI cases that equal mild or prodromal AD. Most MCI trials were negative. Why? One of the key reasons, in my opinion, is that the conversion rates to Alzheimer disease were highly variable.
“The step after that is prevention of AD: that is why we are here today. Our current knowledge of potential prevention of AD comes primarily from epidemiological and long-term observational studies. One of the challenges with trying to prevent dominantly inherited AD is that we only know an average age of onset in each family but not when any given person would develop clinical symptoms. This becomes very important.
“What are the key considerations in trial design:
Who is going to be in it?
How to conduct it?
What are the endpoints?
“I’d say possible inclusion criteria for DIAD could be these: mutation carriers, age, or age relative to average age of onset for specific mutation, MRI status of change, biomarker status, PIB retention, metabolic imaging, clinical status, or combinations of the above. Each of these has problems associated with it, and we could talk about these at length.
“Then there is the complex issue of enrolling carrier versus non-carrier. How could it work if you want to be in the study but do not want to know your carrier status? One option, to enroll non-carriers in an active treatment arm, might be considered unethical. Do we know the drug is safe enough to give to non-carriers for a long time? Alternatively, individuals wishing to stay blinded can be given placebo if confidential testing shows they are non-carriers, and active drug or placebo if they are carriers.
“There are safety considerations. For example, by definition the population is presymptomatic. Risk tolerance is a complex issue for this patient group and must be assessed by the steering group, IRBs, and at the level of patients’ personal consent. Personally, I think the patients and carriers have a huge stake in this decision and should be granted autonomy to have a say in how much risk they are willing to take. It is a very individual-specific issue. Each agent has its own potential safely and efficacy issues.
“There are also pragmatic versus ethical issues. For example, for enrollment and power analysis, you need a specific age group to assess the treatment, but can you fairly exclude people at the fringes of that age group? Do you want to divide by ApoE status, MRI, PIB, or CSF? That will divide the sample into even smaller groups.
“Because of the pragmatic issues, I believe it is going to be fine to look at biomarker changes first and at clinical outcomes second. That is a very big discussion to be had, but this is my feeling about it. You could see biomarker changes in weeks to months, but the clinical outcome in a prevention study could take two to five years or longer.
“Which treatment should be considered? I can’t help but be biased here. That said, I would suggest those with at least Phase 2 safety data to have enough patient exposure to reasonably calculate their safety profile. Agents that have evidence of modifying the pathology of AD should have priority because these agents will most likely impact the most advanced biomarkers. These biomarkers will likely be impacted before clinical manifestation of the disease, and biomarker and imaging agents have greater statistical power of demonstrating a benefit with a small population.
“Should we require known efficacy in mild to moderate AD to consider a specific treatment for DIAN? Results either way may not be applicable to presymptomatic AD.
“Here’s a list of possible classes of treatment to be considered for prevention of DIAD: γ-secretase inhibitors, Aβ aggregation inhibitors, Aβ vaccines, Aβ passive immunotherapies, other neurotrophic compounds (p75). We work on those and have compounds on the way. But there are also cholinergic agents, NMDA antagonists, other neurotransmitter-related compounds, mitochondria-active compounds, tau-targeting compounds. We do not work on those.
“Some detail about ours:
γ-secretase inhibitors: Quite a few are in the clinic. We have a Phase 1 compound that is selective for APP over Notch. I personally think this class of compound is complex when it comes to DIAD because not every presenilin mutant may respond the same way. Our Phase 1 inhibitor has a therapeutic index of more than 65; future compounds further back in the pipeline are even higher. This bigger safety window enables a higher dose.
ELND005, or scyllo-inositol: This anti-aggregation inhibitor blocks the toxicity of Aβ oligomers. It is in Phase 2 for mild to moderate AD, but we have no clinical information on that trial yet. In all honesty, this is one of the few compounds that I think is very, very safe thus far. On the downside, we have not yet established that it is modulating biomarkers. With our γ-secretase inhibitor we do know that it lowers CSF Aβ42.
Immunotherapy: There are at least 14 to 15 different versions of active and passive immunotherapy trials in the clinic worldwide by now. There are many approaches, many sponsors, many clinical investigators working on this. We have learned a great deal. Immunotherapy can achieve real plaque reduction, and CSF tau reduction. But we do not know definitively what that means clinically in Phase 3 studies. The safety issues we ran into with AN1792 are not repeating themselves in newer trials, ours and those of others.
Bapineuzumab Phase 2 findings: The primary efficacy endpoints were not achieved, but post-hoc analyses of completers showed they had a cognitive benefit in that they declined less than the placebo-treated patients. The ApoE4 non-carriers seem to benefit more, so E4 might affect the treatment response. That has come up with several different drugs by now. This is why in Phase 3, separate studies are being conducted with Bapineuzumab in ApoE4 carriers and non-carriers. We have early data to say that Bapineuzumab might reduce CSF tau. On safety, the vasogenic edema is more common in treated than placebo groups; sometimes it is symptomatic. On the other side effects, the percentage of patients with serious adverse events was similar to the percentage of them in placebo. In the Phase 2 study, there were three deaths in the treatment groups that were judged as not associated with treatment.
“I can’t say which treatment should be chosen. But I suggest that the mechanism of action should be known, I do suggest using something related to Aβ, and I suggest it should have a demonstrated effect on biomarkers.
“On endpoints: I suggest choosing biomarker and/or imaging as primary endpoints, and clinical outcome as the secondary endpoints.
“How long should such a study be? The biomarker piece could take 12 to 18 months, the clinical piece three to five years. A five-year study seems reasonable.”
For more on the issue of AD prevention trials, see ARF essay and Ringman et al., 2009).—Edited by Gabrielle Strobel.
This is Part 7 of a seven-part series on presymptomatic detection. See also Parts 1, 2, 3, 4, 5, and 6.

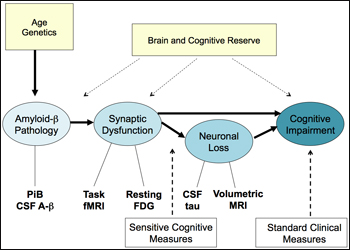
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.