CONFERENCE COVERAGE SERIES
Human Amyloid Imaging 2010
Toronto, Ontario, Canada
09 April 2010
CONFERENCE COVERAGE SERIES
Toronto, Ontario, Canada
09 April 2010
Call it development on fast-forward. Since the first major paper on human amyloid imaging appeared in the scientific literature (Klunk et al., 2004), a burgeoning field of research has sprung up and a recent conference illustrated how it has sped from infancy to adolescence in six years. Sensing an opportunity, multiple diagnostic imaging and pharma companies got into the act even as academic researchers the world over adopted this nascent technique. At this point, researchers are exploring myriad scientific questions about how to visualize the amyloid pathology in a living person’s brain and how to interpret what they see. For their part, commercial players are racing to satisfy FDA requirements for market approval of a slew of candidate ligands. The 4th annual Human Amyloid Imaging (HAI) Conference, held on 8 April 2010 in Toronto, Canada, showcased this dual message clearly. Organized largely by Keith Johnson of Massachusetts General Hospital, with help from Bill Jagust of University of California, Berkeley, and Bill Klunk and Chet Mathis of the University of Pittsburgh Medical School, this meeting drew some 160 attendees from academia and industry worldwide for a day of short talks, two keynote lectures, and the ample, freewheeling discussion that is a mark of distinction for this conference.
One the one hand, there seemed to be no doubt in the room that amyloid imaging works. That is, the main ligands in use—six at present by this writer’s count—all reliably and reasonably specifically image β amyloid deposits in the brain. There was a shared sense that four different commercial F18-labeled ligands, each of which is at a different point in the clinical development pipeline, overall appear to perform quite similarly. And in a sign that the field is beginning to mature, the how-to debate has shifted to smaller methodological issues. Investigators now see the need to work out a degree of standardization on the fine points of data acquisition and analysis so they can better compare findings from study to study. The twin excitement here lies in readying amyloid imaging for robust, larger-scale application in clinical trials as well as to support an earlier, biomarker-driven diagnosis of Alzheimer disease.
On the other hand, the use of these amyloid imaging agents for scientific exploration of the aging brain’s underlying biology has merely begun to scratch the surface. Here, the excitement lies in peeling back a deeper layer of the brain’s mysteries and discovering something fundamentally new. Scientists are increasingly combining amyloid imaging with other forms of measurement in cognitively normal people, such as paper-and-pencil tests, several different modes of brain imaging, AD risk genes, even indicators of cardiovascular health such as blood pressure and vascular amyloid. They do this to address the question of what makes some aging people succumb to the presence of β amyloid in their brains while others can “live at peace with their amyloid for many years,” as Sperling put it. Scientists are hoping that this effort will eventually explain the pathophysiology of AD. On this front, scientists agreed, the field is only just getting underway. Hence, it is at present producing tantalizing but discrepant results that need resolution.
A presentation on this topic won the HAI conference’s $500 Young Investigator Award. Alex Becker, a research fellow at Massachusetts General Hospital, Boston, took a crack at prying apart the factors that together might determine how well aging people can tolerate amyloid accumulating in their brains, and how metabolism is affected by increasing levels of amyloid. Becker measured FDG metabolism, an indicator of how much glucose the brain uses (i.e., how active it is or how hard it is working). He did so in 77 cognitively normal people and focused in on the 21 among them who were amyloid positive. As a group, their metabolism was reduced compared to the amyloid-negative group in cortical areas that form the default mode network, but Becker saw a lot of individual variability. Clearly, other factors were playing a role. To unmask those, Becker first looked at ApoE status. He found that the ApoE4 carriers exhibited a steeper decline in FDG metabolism with advancing age than did the non-carriers. This was region-dependent and primarily the case in frontal regions. Intriguingly, younger ApoE4 carriers started out with higher FDG uptake in certain brain regions than older carriers, even if both had equal amounts of amyloid, and then declined faster.
If ApoE accounted for only a part of the scatter in the initial data, what else was going on? Becker also studied cognitive reserve. He did so in two ways, expressed by way of education and of AMNART scores. (The AMNART is an intelligence test that was designed to be somewhat resistant to the effects of aging.) In this analysis, FDG metabolism went up along with intelligence scores and education in amyloid-negative people, as might be expected. But in amyloid-positive people, Becker found the opposite: FDG metabolism was lower in people with high intelligence/more education. “That is why we did not see this in the pooled group,” Becker told the audience, referring to the lack of a significant relationship between FDG and AMNART in the full group versus the split group. This might suggest that people with high cognitive reserve remain outwardly cognitively normal even though their brain metabolism is already declining.
Along similar lines, Alexander Drzezga of the Technical University in Munich, Germany, reported on his fMRI study of resting-state connectivity in 12 PIB-negative and 12 PIB-positive cognitively normal people as well as 13 PIB-positive people with mild cognitive impairment (MCI). This study detected early signs that the connections between cortical “hubs” in the brain are already beginning to break down in cognitively normal people who were amyloid-positive. This fMRI connectivity loss overlapped with the reduced glucose metabolism that Becker had analyzed. Reisa Sperling of Brigham and Women’s Hospital, Boston, added a cognitive testing angle to this emerging story. Her group’s new analysis of the normal control group in the florbetapir Phase 2 trial suggests that even within what is considered the normal range of an episodic memory test, a higher level of amyloid deposition was associated with lower performance. Overall, the talks generated the impression that amyloid imaging in cognitively normal people is beginning to correlate well with other imaging modalities, and that a larger picture is emerging of subtle deficits across a broad range of indicators in outwardly normal people who have amyloid in their brains. The scientists agreed that more research in larger samples is needed to fill in this emerging picture.
Beyond genetics and cognitive reserve, Vladimir Hachinski, a stroke expert at University of Western Ontario, London, Canada, pleaded with his amyloid imaging colleagues to take vascular disease into consideration. Instead of being excluded from AD studies, people with vascular disease should be made a focus of study because “when you have vascular disease, that is when amyloid causes cognitive impairment,” Hachinski said (see also MCI conference). This view drew widespread agreement. (Interested to learn more? See upcoming Cerebral Amyloid Angiopathy Conference.) Likewise, the day heard repeated calls for imaging inflammation as a modifying factor, though no one appeared to be aware of suitable ligands beyond 11C PK11195, which has not found widespread acceptance.
The HAI Conference saw intense discussion about the technical challenges amyloid imaging is confronting at present. They seemed solvable. Above all, scientists need to agree on which brain area to use as a normalization region (see Part 2 of this series) and on how to draw its contours, so that studies at different sites become more comparable. Scientists shared their troubles with a technical problem called partial volume effects, and discussed whether to try to tame it with what’s called a CSF correction. The correction measures more amyloid in atrophic brains but less so in non-atrophic brains. Many scientists agreed that it is best to process the data with and without this correction and evaluate what the correction does to the outcome.
Speakers and audience members also argued about which analysis methods work best for particular goals, i.e., multicenter drug studies versus academic explorations of exactly where an amyloid scan becomes “positive” along a poorly understood process of accumulation. They considered different ways of setting cutoffs for this transition. They agonized over how they can much more rigorously define what it means that someone is “cognitively normal.” This might lessen a selection bias they suspect of being at the root of discordant results in current research of cognitively normal people, which are causing some confusion at this early stage of research. “Nobody goes to bed on Monday and wakes up impaired on Tuesday,” said Cliff Jack of the Mayo Clinic in Rochester, Minnesota. “It is a matter of where on the continuum you draw your sample. Most samples are small, most have biases, and that explains the differences between the studies out there.” (For more on these issues, see Part 2 of this series.)
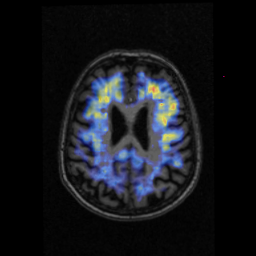
PIB-PET From a Clinically Normal 80-Year-Old Woman
Her mean cortical PIB is below threshold, but she has focal deposition (yellow-red). Many normal subjects have clear focal uptake but have average cortical uptake that is insufficient to categorize them as amyloid-positive. Longitudinal studies will tell if this uptake accumulates over time. Image credit: Keith Johnson, Massachusetts General Hospital
In contrast, one finding stood out for its clarity throughout the day. With great consistency, the ApoE4 AD risk allele shows up in the AD-prone group of very mildly impaired or cognitively normal volunteers who have amyloid. Mark Mintun of Washington University, St Louis, Missouri, showed how ApoE4 carriers trend upwards in their amyloid load at younger ages than non-carriers; Elizabeth Mormino of the University of California, Berkeley, found ApoE4 carriers overrepresented among the amyloid-positive subgroup in her study of cognitively normal volunteers. Kenji Ishii of the Tokyo Metropolitan Institute of Gerontology, Japan, found this link in the J-ADNI cohort, as well; there, all ApoE4-carrying AD and MCI patients to date were amyloid-positive, as were half the ApoE4-carrying cognitively normal participants. Osama Sabri of the University of Leipzig, Germany, presented data on a link between carrying an ApoE4 allele and having brain amyloid as measured by the ligand florbetaben; in this study, uptake of the amyloid ligand went up with each copy of the allele in AD patients.
The poster session deepened this impression. Christopher van Dyck at Yale University School of Medicine in New Haven, Connecticut, tested in his group’s own study population the previous finding that ApoE4 drives preclinical amyloid deposition in a dose-dependent way (Reiman et al., 2009). The Yale investigators ran PIB-PET scans in 270 cognitively normal volunteers in their fifties and sixties who had a first-degree relative with Alzheimer’s, and confirmed that in their sample, too, ApoE4 carriers had considerably more amyloid than non-carriers of the same age, sex, and education. Amyloid-positive volunteers tended to be slightly weaker on tests of episodic memory. Shizuo Hatashita of Shonan Hospital in Atsugi, Japan, reported that among 34 people with MCI who received a PIB scan and were followed clinically for up to two years, the PIB-positive ApoE4 carriers progressed faster to meet an AD diagnosis.
These new data jibe with recent published reports suggesting that ApoE4 carriers tend to accumulate more brain amyloid than non-carriers (e.g., Drzezga et al., 2009), do so at younger ages (Morris et al., 2010), age with reduced blood flow in the brain (Thambisetty et al., 2010), and have lower CSF Aβ42 levels in various biomarker studies including ADNI. “The ApoE effect on amyloid deposition seems incredibly concordant,” observed Neil Buckholtz of the National Institute on Aging. Buckholtz qualified, however, as did other scientists, that this seemingly definitive conclusion might yet shift if independent research confirms a paper claiming that some of the ApoE effect on age of disease onset is actually the doing of the nearby gene TOMM40 (Roses et al., 2009; Lutz et al., 2010).
Beyond implicating ApoE4 in amyloid deposition, researchers at HAI exchanged news on various fronts. For example, Ishii presented the first amyloid imaging results from the Japanese ADNI (J-ADNI) study. In brief, J-ADNI is going well, Ishii said, having enrolled 354 out of the desired 600 participants as of this month. Thirteen amyloid imaging sites to date have imaged 21 people with AD, 28 with MCI, and 46 controls aged 66 to 74. Of those, 95, 75, and 24 percent, respectively, have proven amyloid-positive. Overall, J-ADNI’s visual assessment performed as well as the quantitative one, but small amounts of amyloid in borderline positive cases are better detected with dynamic data acquisition and DVR (see Part 2), Ishii said.
First data on epidemiological modeling of brain amyloid in the elderly population were on the menu (see Part 2), as well as Phase 3 autopsy validation for the 18F ligand florbetapir (see Part 3). Also new were Phase 2 data on two other 18F ligands (see Part 4). First human data on a fourth ligand, which particularly intrigued some imagers, were presented at the Springfield Conference in Geneva last month. To learn all about that, see Part 5.—Gabrielle Strobel.
This is Part 1 of a six-part series. View a PDF of Part 1. See also Part 2, Part 3, Part 4, Part 5, Part 6. View PDF of the entire series.
At the 4th Human Amyloid Imaging Conference held on 9 April 2010 in Toronto, several themes generated intense discussion. On the technical side, the question kept cropping up of what is the right reference region to use in amyloid imaging. Typically, imagers compare any given region’s amyloid burden to that of cerebellum, because this brain area remains relatively unscathed deep into AD progression. Trouble is, it doesn’t always. In some studies, some parts of the cerebellum do show some amyloid. For example, this is the case in autosomal-dominant forms of AD and must be taken into account for amyloid imaging studies of the DIAN, noted Stephen Salloway of Butler Hospital in Providence, Rhode Island. In fact, at HAI, scientists led by David Brooks at Hammersmith Hospital in London presented first PIB imaging data on seven presenilin-1 mutation carriers along with those of 10 sporadic cases and 10 controls. (These are U.K. families in the care of Martin Rossor and Nick Fox at University College, London.) This study indeed found increased cerebellar uptake in some mutation carriers.
Some studies have bracketed off amyloid-containing sub-areas of the cerebellum and use the rest as the reference region, while other studies use the entire cerebellum. If scientists optimize the reference region they choose, they can make their data look stronger by widening the separation between amyloid-positive and amyloid-negative groups. At a different conference last month in Geneva, Eric Reiman of the Banner Alzheimer’s Institute in Phoenix, Arizona, illustrated this point. He showed on a slide how comparing a given dataset against a reference region drawn from different parts of the cerebellum shifted uptake values for the brain regions of interest considerably.
In Toronto, several speakers emphasized the need to find consensus not only on which reference region to use but, even more so, on precisely how to delineate it on the brain atlas. This delineation is important because it affects the threshold above which a person is judged to be amyloid-positive. Different groups at present draw this cutoff in different ways, making comparisons difficult. “The cutoffs are a bit fuzzy,” said Val Lowe of Mayo Clinic in Rochester, Minnesota. “We are not always comparing apples to apples yet. To do that, we need to agree on what reference region to use,” agreed Jessica Langbaum, also at Banner. Other scientists cautioned that shrinkage over time of the cerebellum could introduce error into longitudinal studies, requiring its own correction. Atrophy in general is a bit of a puzzle to amyloid imagers. They don’t know if a person loses plaque as they lose brain tissue, or if the plaques stay and become more concentrated, noted Bill Klunk of the University of Pittsburgh, Pennsylvania.
Scientists led by Brooks presented two posters describing how they compared the cerebellum’s and the pons’s discriminatory power as reference regions for a PIB and an 18F flutemetamol dataset (18F flutemetamol is a ligand developed by GE Healthcare). They concluded the pons is suitable in studies where cerebellum is not, but discussion reached no general consensus on the issue. Other scientists, for example, Osama Sabri from the University of Leipzig, Germany, showed data supporting the use of the cerebellar cortex for reference.
Beyond normalization, technical debate also revolved around the methods by which neuroimagers analyze the raw data in order to quantify the amyloid in a person’s brain. For example, scientists discussed the relative merits of a method called distribution volume ratio (DVR) versus one called standard uptake value ratio (SUVR). Without getting overly arcane in a news story, the gist of the argument is that the DVR method is generally considered to be more sensitive, but as the field is expanding, a growing number of investigators use only the simpler SUVR method. DVR requires a data acquisition lasting 60-90 minutes while the PET camera makes a movie of about 50 frames. The SUVR comes from an image that is summed over 10-30 minutes, so the participant lies in the scanner for a shorter period of time.
At HAI, Ann Cohen from the University of Pittsburgh evaluated both methods side-by-side in 62 cognitively normal controls. The question was not whether the SUVR can distinguish garden-variety AD cases from controls—by general consensus, it can—but how sensitive it is in picking up small amounts of amyloid in people whose levels might hover right around a threshold of amyloid positivity. For these people, subtle differences in the analysis method could well determine on which side of such a cutoff they end up, hence, changing a study’s result. Cohen reported that the DVR produced data within a narrower range than the SUVR method and classified three people as amyloid-positive whom the SUVR did not pick up as such. Kenji Ishii of the Tokyo Metropolitan Institute of Gerontology in Japan, in presenting the first amyloid imaging results from the J-ADNI study (see Part 1 of this series), concurred, saying that DVR analysis showed considerable amyloid deposition in three of 33 people classified as negative by the SUVR. Others disagreed. Sabri noted that his proof-of-mechanism study of the Bayer Healthcare F18 compound florbetaben found no such difference.
In fact, sensitivity was a sensitive subject throughout the day. DVR versus SUVR represents but one aspect of it; another is whether to measure amyloid in the brain globally or by region. For her part, Cohen reported that among the 62 volunteers, regional PIB values classified a greater number as amyloid-positive than did global PIB values. She suggested that a global analysis does a fine job of analyzing widely distributed amyloid deposition, but not of measuring the first indication of amyloid in any brain region. Elizabeth Mormino of the University of California, Berkeley, agreed, saying that in her hands, too, the earliest increases in amyloid most reliably lit up in certain local regions, above all the precuneus. For his part, Keith Johnson of Massachusetts General Hospital, Boston, suggested that this distinction is scientifically interesting beyond the immediate goal of getting amyloid imaging ligands approved. “In terms of detecting amyloid, we have learned something in the past year. All of us have seen cases where there is highly focused uptake in small regions, and we follow these people and see it spread from there. I hypothesize that these people have a sea of prefibrillar amyloid in their brain, and certain areas ‘poke through’ with fibrillar deposition and reveal themselves. In detecting amyloid, which is the FDA requirement we are all thinking about, is it important to recognize these biological features, or just the instrumental features we are talking about?”
Similarly, Klunk recommended that investigators stay acutely aware of the sensitivity of the methods they choose. “We are beginning to see amyloid-negative cases with neuropathologically evident amyloid, suggesting that the threshold we use to detect reflects a fair amount of amyloid in the brain. That is true even with the most sensitive DVR measures. If we miss more by using SUVR to measure amyloid, do so globally, and miss even more by using an F18 ligand with higher white matter staining, then we may end up missing quite a lot.” Scientists said this conundrum represents the tension between scientific accuracy and the practical and financial constraints of broadly applicable multicenter protocols. In short, investigators need to select their methods based on the sensitivity needed for each particular study.
One good example for the importance of analytic sensitivity is this key research question: What is a positive PIB scan? “Answering this simple question is not straightforward at all,” said Mormino. In the past two years, one of the greatest surprises in the field has been the large proportion of cognitively normal people who have significant amounts of amyloid in their brains. And yet, defining just from what point on a PIB scan counts as positive is tricky. At HAI, Mormino presented a new study to get at this problem. She compared two methods of establishing a threshold—one a previously published objective approach that removes outliers from among elderly control data (Aizenstein et al., 2008), and a subjective, simple approach of cutting at two standard deviations above the mean PIB value she’d measured in seven twentysomething controls. Mormino then applied the cutoffs derived in these two ways to 52 old controls and 25 AD patients.
The result? Both methods classified all AD patients as PIB-positive. Among the elderly controls, the Aizenstein method proved more conservative, putting eight of 52 into the PIB-positive bucket versus 15 as per the subjective approach. These eight folks are probably truly positive, validating this approach to creating a cutoff, Mormino said. They also tend to be older, more likely to have ApoE4, have smaller hippocampi and worse episodic memory than the people in the PIB-negative group; hence, their profile fits a preclinical AD picture. The seven volunteers that came out as PIB-positive by one approach but not the other form an interesting group that warrants longitudinal follow-up with imaging and other preclinical measures, Mormino said. Their signal could be noise, or it could be biologically meaningful, representing an earlier stage of amyloid accumulation. As to sensitivity, for this study Mormino chose PIB as the ligand and DVR as the analysis method.
HAI attendees agreed that it is important to forge some agreement around exactly what constitutes amyloid-positive, and also, what constitutes cognitively normal. Why? These definitions will influence much future research. For example, researchers are beginning to experiment with modeling PIB data from cognitively normal people to generate initial incidence and prevalence estimates. Mark Mintun showed how that could work with data from volunteers at Washington University, St. Louis. He ran longitudinal PIB-PET scans on 129 people age 45 to 88, set a threshold, and calculated how quickly people crossed it. From there, Mintun computed an incidence rate of 2.9 percent of adults becoming amyloid-positive per year. The prevalence of having plaques in the brain came out as increasing from 4.4 percent in one’s fifties to 14 percent in the sixties and 50 percent in one’s nineties. Mintun calculated a delay between amyloid plaques and dementia of about a decade. This points to a future epidemiology of brain amyloid in the normal population. Yet in these early days, scientists operate with small datasets and with assumptions that vary somewhat from site to site; hence, their results also vary from site to site.
Illustrating this, Chris Rowe of Austin Hospital in Melbourne, offered the perspective from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging, which also supports epidemiological calculations (Ellis et al., 2009). His calculation of delay between plaques and dementia came out closer to 20 years. In general, Rowe had even more “depressing” numbers, as he called them. “In our hands, the prevalence of amyloid positivity in those over eighty is 50 percent. We have plenty of old people in the study, and it looks as if amyloid shows up in everyone if you live long enough,” Rowe said. In discussion, suggestions for this extreme variation in the numbers ranged from the facetious (“watch that kangaroo meat—maybe it’s amyloidogenic”) to the serious. The argument that echoed throughout the day was that results depend on how the “normal” group is being assembled. In the WashU study, participants are assessed every year and moved out of the “normal” pool as soon as they show deficits, whereas in the AIBL study, the normal cohort might contain more people with mild impairments, some said. Bill Jagust of the University of California, Berkeley, concluded the topic with a call on neuroimagers everywhere to insist on careful characterization of the normal cohort in their future studies.—Gabrielle Strobel.
This is Part 2 of a six-part series. View a PDF of Part 2. See also Part 1, Part 3, Part 4, Part 5, Part 6. View PDF of the entire series.
No Available Comments
At present, at least four companies are developing amyloid imaging agents coupled to the radiotracer 18F rather than 11C. While the original ligand 11C PIB is still considered to be the most sensitive of the amyloid imaging options to date, an 18F-labeled compound must be had if amyloid imaging is to become widely used in large-scale multicenter drug testing and earlier-stage diagnosis. At the 4th Human Amyloid Imaging Conference, held 9 April 2010 in Toronto, Canada, scientists presented data on three of the 18F contenders; a fourth was discussed at the 11th International Geneva/Springfield Symposium on Advances in Alzheimer Therapy held earlier this spring in Geneva, Switzerland. Counting by clinical milestones, Avid Radiopharmaceuticals’ florbetapir (formerly known as 18F-AV-45) is arguably furthest ahead in the race to FDA qualification. Having been formally studied in some 270 people to date, it supplies the ADNI-GO study and is the ligand of choice for the ADNI 2 grant. At HAI, Adam Fleisher of the Banner Alzheimer’s Institute in Phoenix, Arizona, presented an interim analysis of the first six cases of a Phase 3 histopathology validation study that the FDA requires as part of the qualification package for each ligand. Phase 2 results on florbetapir were reported at the 2009 ICAD Conference in Vienna, Austria (see ARF related ICAD story). (For Phase 2 and Phase 1 data on the other three 18F ligands, see Part 4 and Part 5 of this series.)
This is a 26-center U.S. study involving 150 adults at the end of their lives. Approximately half have AD; all have fewer than six months to live, many of them in hospice. They undergo a 10-minute florbetapir scan and some neuropsychological testing, if possible, and are then followed until autopsy. The basic idea is to compare the accuracy of a visual rating of florbetapir imaging by three trained readers and a quantitative analysis by SUVR of six brain regions to the participant’s subsequent postmortem pathology as measured conventionally by immunohistochemical amyloid burden and CERAD scoring of plaque density. The study was funded by AVID; Fleisher is a site investigator at Banner on this trial, with Banner Sun Health Research Institute acting as the core pathology laboratory. Fleisher reports no personal financial relationship with the company.
The first patient to be autopsied in this study was a 47-year-old woman who met clinical diagnostic criteria for MCI and scored 24 on the MMSE. She had end-stage kidney disease and died 11 days after the scan. She was amyloid-negative on the visual read and by SUVR quantification, and her brain contained no amyloid pathology. Her cognitive impairment might have been a result of her dying from kidney failure, Fleisher speculated. The second patient was an 82-year-old man with a clinical AD diagnosis and an MMSE of 14. He had metastatic prostate cancer and died 53 days after the scan. His brain amyloid was below threshold both by the florbetapir visual read and quantification and by way of postmortem pathology. He did have isolated neurofibrillary tangles in his medial temporal lobes, Fleisher noted. The third patient was a 78-year-old man diagnosed clinically with advanced Parkinson disease dementia (PDD); he was bedridden, rigid, scored 5 on the MMSE, and passed away the day after his florbetapir scan. This man was amyloid-positive by florbetapir imaging, and postmortem pathology yielded an AD diagnosis with plaques, tangles, and diffuse cortical Lewy bodies. The fourth, fifth, and sixth patients, aged 76 to 84, all had clinical diagnoses of AD with MMSE scores of 6 to 0. All died within a month after the scan of their end-stage AD, and all were amyloid-positive by both imaging measures as well as by both postmortem pathology measures.
A larger sample size is necessary before the investigators can determine how sensitive florbetapir is compared to postmortem pathology, Fleisher said. The common thread so far is that, in every case, the amyloid imaging measure matched up with postmortem pathology, even if those two together contradicted the prior clinical diagnosis. This suggests that florbetapir retention actually measures amyloid pathology, Fleisher said. Others agreed, noting that until now there had been some lingering doubt whether these ligands really “see” amyloid. “But now postmortem validation is coming on a larger scale beyond individual case studies. If it shows, as it did here, that the cases themselves vary greatly but imaging and pathology always correlate, then that counts as proof and hopefully will lead to regulatory approval,” commented Alexander Drzezga, a neuroimaging expert at the Technical University in Munich, Germany, who is not involved in these hospice studies.
That the visual read came out in line with the quantitative assessment implies that amyloid imaging eventually may be read rather simply at local PET centers, neurologists’ offices, and in large multicenter drug trials. One worry had been that it might require highly trained, rare specialists or quantitative analysis. This is a priority in implementing any new imaging procedure on a broad basis. “The visual impression of an amyloid PET scan is most important at the sites,” agreed Osama Sabri of the University of Leipzig, Germany, who tests a competing ligand in Phase 2 and an ongoing Phase 3 histopathology validation (see Part 4).
Fleisher’s talk generated a fair amount of praise and hallway buzz throughout the day. One point of criticism came up as well. It is that the investigators did not also take an MRI to prove that the anatomic regions seen in the PET scan registered exactly with the areas later studied by postmortem pathology. Some critics pointed out that the spatial resolution of amyloid PET is too low to align regions precisely without an accompanying MRI scan. Others countered that while that is true, hospice studies are ethically sensitive as it is, and lessening the burden on a person who is near death rightly takes precedence. “I would hope that as long as you have global amyloid positivity and an approximate registration, it will prove the point to the FDA well enough,” said Drzezga.
Case-by-case histopathology research is continuing within academic research as well. At HAI, Val Lowe from the Mayo Clinic in Rochester, Minnesota, added new examples to the small existing literature (Ikonomovic et al., 2008; Bacskai et al., 2007; Leinonen et al., 2008; Burack et al., 2010; Cairns et al., 2009; Rosen et al., 2010). Overall, Lowe found increased PIB binding in three people who had been clinically diagnosed as having Alzheimer disease, amnestic mild cognitive impairment (aMCI), and dementia with Lewy bodies (DLB), respectively; all three also had corresponding pathology according to CERAD criteria. A person who died with a clinical diagnosis of non-amnestic MCI and a normal control did not. One of the finer points of this study, though, for example, which kinds of plaques prevailed in the respective cases, Lowe noted seeing considerable variation.—Gabrielle Strobel.
This is Part 3 of a five-part series. View a PDF of Part 3. See also Part 1, Part 2, Part 4, Part 5, Part 6. View PDF of the entire series.
No Available Comments
At the 4th Human Amyloid Imaging (HAI) Conference held 9 April 2010 in Toronto, Canada, one 18F amyloid ligand strutted its Phase 3 stuff, while two others flaunted Phase 2 results. A third ligand was fresh in people’s memory from a recent presentation of Phase 1 (see Part 5).
The Bayer Healthcare Ligand
Osama Sabri, University of Leipzig, Germany, is the principal investigator of the Phase 2 clinical trial program for Bayer Healthcare’s ligand, 18F florbetaben. Bayer funded this study; Sabri has received consulting fees from the company. For those of our esteemed readers who are trying to keep up with the alphabet soup that is amyloid tracer terminology, florbetaben is a stilbene compound formerly known as BAY94-9172 or AV-1/ZK. It differs from Avid’s florbetapir (see Part 3 of this series) only by a carbon-to-nitrogen substitution in one position. Bayer Schering Pharma licensed it from AVID and began testing it in single-site studies, first at Austin Hospital in Melbourne, Australia, led by Chris Rowe (Rowe et al., 2008), then in Leipzig by Sabri. In Toronto, Sabri presented a full analysis of an 18-center Phase 2 study conducted in Australia, the U.S., and Europe. It probed florbetaben’s ability to distinguish AD from normal in 150 participants aged 55 and older who underwent PET and MRI scans.
Sabri emphasized that this trial’s mark of distinction is its combination of assurance that the control participants are indeed cognitively normal (which resulted in a comparatively low amyloid-positive rate in the control group), with external quality control of each image at the Molecular Neuroimaging Institute, a service company in New Haven co-founded by Kenneth Marek and John Seibyl. “Every image was evaluable, we had no dropouts due to quality. That is unusual for an 18-center study,” Sabri said.
The sensitivity and specificity with which visual assessment of the scans by three blinded readers replicated the clinical diagnosis served as the primary endpoint of this trial. That’s because Bayer intends eventually to market florbetaben as a diagnostic aid and therefore wants it to be clinically practicable on-site, Sabri said. An automated quantification of the amyloid signal served as the secondary endpoint; this was done by obtaining standard uptake value ratios (SUVRs) of volumes of interest derived by gray matter segmentation of the participants’ MR scans. The segmentation served to diminish the effect of florbetaben’s non-specific white matter binding, Sabri said. As with quality control of the images, the New Haven group performed this quantification as an external site for the entire study.
The primary endpoint came out as 80 percent sensitivity, 90 percent specificity on visual read. For a trial of this size, this is but a small decrement from the higher results of prior single-site studies on florbetaben or smaller multicenter studies with 18F compounds, Sabri emphasized, insisting, “These numbers are great.” In this study, the clinical diagnosis served as the standard of truth.
In the secondary endpoint, each of eight regions of interest showed a highly significant difference between the AD patients and healthy volunteers. The posterior cingulate distinguished particularly well, Sabri said.
The groups had some overlap: six healthy volunteers lit up as amyloid-positive and 16 patients with diagnosed AD came out as amyloid-negative. The next study, a Phase 2b, will enroll 270 participants, and a histopathology validation study is also currently underway, Sabri added. For a diagram of this compound’s structure and its reported affinity to AD brain homogenate (see ARF related news story).
The GE Healthcare Ligand
Rik Vandenberghe at the University Hospital Leuven, Belgium, presented for the first time the primary outcome analysis of a smaller multicenter Phase 2 trial of an 18F-labeled derivative of PIB called 18F-flutemetamol, formerly known as 18F AH110690. This follows a Phase 1 study with 16 patients (Nelissen et al., 2009). GE Healthcare funds this research.
This trial took place at seven sites in Belgium, Denmark, and Sweden. It enrolled 27 AD patients, 20 people with MCI, 15 cognitively normal controls above the age of 55, and 10 younger than that. Like in the florbetaben trial, the investigators wanted to know primarily how well a visual assessment of the images obtained with this ligand distinguished probable AD from the elderly controls. On this goal, five blinded readers categorized 25 of the 27 as having “raised” uptake, and 14 of the 15 cognitively normal elderly as “normal,” yielding a sensitivity and specificity of 93 percent. The raters’ independent classifications generally agreed with each other, Vandenberghe said. As in the florbetaben study, the visual read was concordant with a quantitative SUVR analysis. All told, flutemetamol worked in this study; next, an ongoing study is going to calculate how strongly flutemetamol binding predicts AD in an MCI population, Vandenberghe said.
Next, the scientists asked how this compares with PIB, widely seen as the gold standard in amyloid imaging. They ran a small comparison of PIB and flutemetamol. Incidentally, numerous academic investigators over the years have called for side-by-side comparisons of the different amyloid imaging ligands. An AstraZeneca representative casually told this reporter earlier this year that her company was willing to make their compound available for direct comparison, perhaps because it seems promising but is running behind the others at present (see Part 5 of this series). But in general, commercial sponsors have shown limited appetite for such an exercise up until now. This small PIB-flutemetamol comparison represents an exception, made easier because GE has also licensed PIB.
In any event, Vandenberghe and colleagues administered both PIB and flutemetamol scans on the same day to 20 AD and 20 MCI patients. Again by visual assessment, the PIB scans were 100 percent concordant with the flutemetamol scans, Vandenberghe told the audience. Both ligands identified all 20 AD and nine of the 20 MCI patients as having raised brain amyloid levels. However, the scientists did not compare the two ligands in the same cognitively normal controls, where subtle differences in their sensitivity might be most likely to show up.
The normal controls in this study received a flutemetamol scan. A small fraction of this group, one out of 15 elderly controls, turned out amyloid-positive than in similar studies with PIB (see Part 2). A larger normal cohort is currently being studied, but Vandenberghe speculated that these differences come down to the cohort. “This is an important question. We find a very high concordance between PIB and flutemetamol. It does not depend on the ligand; it depends on how the cohort is defined and subjects recruited,” Vandenberghe said (see also Part 1 of this series),
In a later discussion about whether 11C PIB still has a place, Vandenberghe noted that his site on occasion had to call back volunteers for a second scan when local production of 11C PIB failed, whereas this snafu did not happen with the 18F compound. “For large multicenter trials, it is simply more practical,” he said. For certain research questions, however, scientists generally agreed that PIB still reigns supreme. “For our new program project, we chose PIB because we are looking for very subtle signals. But I am thrilled that the data on the 18F ligands are so concordant, because we will need large primary prevention trials, and they seem comparable and reliable for that,” said Reisa Sperling of Brigham and Women’s Hospital in Boston (see also Sperling talk from a recent conference in Miami Beach, Florida).—Gabrielle Strobel.
This is Part 4 of a six-part series. View a PDF of Part 4. See also Part 1, Part 2, Part 3, Part 5, Part 6. View PDF of the entire series.
No Available Comments
Below we list the 39 abstracts presented at the Human Amyloid Imaging Conference held 9 April 2010 in Toronto, Canada. All are published in the Alzforum Papers of the Week database. The accompanying news series covers some of these studies, but far from all; hence, this document offers the curious reader a wealth of new data to peruse. The Alzforum editors are grateful to co-organizer Keith Johnson for obtaining permission to publish the abstracts; we also thank all the authors for their generosity in sharing their latest data with the worldwide Alzheimer research community.—Gabrielle Strobel.
Human Amyloid Imaging 2010 Meeting Abstracts
Lowe VJ, Parisi JE, Jack Jr CR, Kantarci K, Senjem ML, Wiste HJ, Weigand SD, Kemp BJ, Petersen RC. Comparison of PiB distribution on PET with beta-amyloid deposits at autopsy Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Fleisher AS, Schneider JA, Beach TG, Bedell BJ, Zehntner SP, Clark CM, Krautkramer MP, Pontecorvo MJ, Joshi A, Skovronsky DM. Update on florbetapir F 18 (18F-AV-45) PET clinical studies. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Mintun MA, Vlassenko AG, Sheline YI, Morris JC. Prevalence and incidence of beta-amyloid accumulation from cross-sectional and longitudinal [11C] PIB PET imaging. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Mormino EC, Hayenga AO, Yen IV, Rabinovici GD, Baker SL, Jagust WJ. Not quite PIB-positive, not quite PIB-negative: low levels of beta-amyloid deposition in elderly, normal control subjects may precede AD-like changes. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Cohen AD, Price JC, Klunk WE, Weissfeld LA, Redfield AS, Berginc M, Rosario BL, Nebes RD, Mathis CA. Comparison of approaches for establishing cut-offs for [C-11] Pittsburgh Compound B. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Sabri O, Gertz H-J, Dresel S, Heuser I, Bartenstein P, Bürger K, Hiemeyer F, Lehr S, Wittemer-Rump S, Barthel H. Multicenter phase 2 trial to test florbetaben for b-amyloid (Ab) brain PET in Alzheimer’s disease (AD). Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Vandenberghe R, Van Laere K, Ivanoiu A, Salmon E, Triau E, Hasselbalch S, Law I, Andersen A, Korner A, Brooks DJ. Primary outcome analysis of the multicentre phase II trial of 18F-flutemetamol, a Pittsburgh Compound B derivative for in vivo beta amyloid imaging. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Sperling RA, Johnson K, Doraiswamy PM, Reiman EM, Sabbagh MN, Sadowsky CH, Carpenter A, Clark CM, Flitter M, Pontecorvo MJ. Amyloid deposition detected with 18F-AV-45 is related to decreased memory performance in clinically normal older individuals. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Rowe CC, Chetelat G, Pike K, Psych D, Jones G, Ellis K, Li Q-X, Martins R, Ames D, Villemagne VL. A comparison of imaging, cognitive and blood biomarkers for prediction of cognitive decline. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Drzezga A, Becker JA, Sreenivasan A, Talukdar T, Van Dijk K, Sullivan C, Schultz AP, Sepulcre J, Buckner RL, Sperling RA. Relation between hypometabolism, impaired functional connectivity and β-amyloid load in pre-dementia stages of Alzheimer’s disease. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Becker JA, Carmasin J, Maye J, Rentz DR, Buckner RL, Sperling RA, Johnson KA. Amyloid deposition and FDG metabolism in relation to age in APOE4 carriers. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Ishii K, Sakata M, Oda K, Ishiwata K, Senda M, Ito K, Kuwano R, Iwatsubo T. The status and the first preliminary results of amyloid imaging in the Japanese Alzheimer’s Disease Neuroimaging Initiative (J-ADNI) study. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Jack Jr CR, Wiste HJ, Vemuri P, Weigand SD, Senjem ML, Bernstein MA, Gunter JL, Petersen RC, Aisen P, Knopman DS. Brain Aβ amyloid measures and MRI are complimentary predictors of progression from MCI to AD. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Nordberg A, Schöll M, Kadir A, Andreasen N, Almkvist O. PET imaging of fibrillar amyloid in brain more sensitive diagnostic marker than CSF Aβ42? Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Morris JC, Roe CM, Grant EA, Holtzman DM, Fagan AM, Mintun MA. PIB Imaging and CSF biomarkers predict cognitive impairment and dementia of the Alzheimer type (DAT). Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Brück A, Locascio JJ, Gomperts SN, Rentz DM, Becker JA, Memole L, Carmasin J, Maye J, Growdon J, Johnson KA. Patterns of amyloid deposition distinguish non-demented Parkinson’s disease from normal aging. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Carmasin JS, Maye JE, Marshall GA, Becker JA, Sperling RA, Rentz DM, Johnson KA. Overestimation of memory performance in normal elderly subjects is associated with amyloid burden in the temporal lobeOverestimation of memory performance in normal elderly subjects is associated with amyloid burden in the temporal lobe. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Carpenter AP, Pontecorvo MJ, Sadowsky C, Hassman HA, Edell S, Clark CM, Hefti F, Joshi A, Burns J, Skovronsky DM. Pharmacokinetics and pharmacodynamics of 18F-AV-45 (florbetapir F 18) PET imaging in Alzheimer’s disease (AD) and healthy control subjects: results of a phase II trial: 18F-AV-45-A03. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Chau WF, Peterson S, Farrar G, Eko-Ebongue S, Barnes C. Evaluation of metabolites of [18F]flutemetamol, an amyloid imaging agent in human and rat in vitro and rat in vivo models. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Chertkow H, Nikelski J, Leger G, Litwin L, Whitehead V, Evans A. Using FDG PET and PIB (Pittsburgh B) PET imaging to distinguish atypical Alzheimer’s disease and fronto-temporal dementia cases. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Edison P, Hinz R, Ramlackhansingh A, Thomas J, Turkheimer FE, Brooks DJ. Can we use pons as a reference region for the analysis of [11C]PIB PET? Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Elmaleh DR, Shoup TM, Johnson K, Selkoe D, Fischman AJ. Evaluation of inositol and benzothiazole derivatives for amyloid-β peptide inhibition and amyloid imaging. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Gomperts SN, Brück A, Locascio JJ, Rentz DM, Becker JA, Memole L, Carmasin J, Sperling RA, Growdon J, Johnson KA. Regional burden of Aβ-amyloid relates to cognitive function in Parkinson’s disease. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Hatashita S, Yamasaki H. Progression of MCI associated with amyloid deposition using PIB PET imaging. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Ichise M, Becker G, Barthel H, Patt M, Luthardt J, Gertz H-J, Schultze-Mosgau M, Rohde B, Reininger C, Sabri O. Kinetic modeling of florbetaben PET data to quantify β-amyloid binding in the human brain. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Ikonomovic MD, Abrahamson EE, Mathis CA, Price JC, Srinivasan S, Debnath ML, Hamilton RL, DeKosky ST, Klunk WE. Histological comparison of neocortical β-amyloid plaque labeling using fluorescent derivatives of flutemetamol (3’-F-PiB) and PiB. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Klunk WE, Snitz BE, Cohen AD, Price JC, Mathis CA, DeKosky ST, Lopez OL, Saxton JA. Comparison of longitudinal changes in amyloid deposition and cerebral metabolism in early onset familial AD. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Langbaum JBS, Chen K, Liu X, Fleisher AS, Reeder S, Bandy D, Alexander GE, Caselli RJ, Reiman EM. Association between pulse pressure and fibrillar amyloidbeta burden in cognitively normal, late middle-aged people at three levels of genetic risk for Alzheimer’s disease. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Larvie M, Van Leemput K, Becker JA, Maye JE, Carmasin JS, Sperling RA, Fischl B, Johnson KA. Hippocampal substructural volume in relation to amyloid deposition. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Maye JE, Carmasin JS, Becker JA, Rentz DM, Sperling RA, Johnson KA. Maternal history of dementia is associated with amyloid deposition in clinically normal older individuals. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Park DC, Rodrigue KM, Kennedy KM, Rieck JR, Hebrank AC, Devous MD. Amyloid burden in normal aging: The Dallas Lifespan Brain Study. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Rabinovici GD, Alkalay A, Marchant NL, DeCarli C, Mungas DM, Chui HC, Reed BR, Jagust WJ. Contribution of amyloid and cerebrovascular disease to cognitive impairment in individuals with high vascular risk. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Ramlackhansingh AF, Knight WD, Okello A, Ryan N, Turkheimer F, De Llano SRM, Edison P, Douglas J, Fox NC, Brooks DJ. Amyloid imaging in presenilin 1 mutation carriers with 11C-PiB PET. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Rentz DM, Becker JA, Frey MT, Carmasin J, Maye J, Olson L, Johnson KA, Sperling RA. A challenging test of name retrieval may be sensitive to early amyloid deposition in normals. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Rinne JO, Koivunen J, Scheinin N, Aalto S, Vahlberg T, Någren K, Parkkola R, Viitanen M. PET imaging of amyloid deposition in patients with mild cognitive impairment: A two year follow-up study. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Sojkova J, Zhou Y, Kraut M, Resnick S, Wong DF. Effects of acquisition time in [11C]PIB PET dynamic studies of non-demented older adults. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Thambisetty M, Tripaldi R, Riddoch-Contreras J, An Y, Campbell J, Sojkova J, Kinsey A, Hye A, Guentert A, Resnick S. Proteome-based identification of plasma biomarkers for brain amyloid burden. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
van Dyck CH, Brück A, Barcelos NM, Benincasa AL, Planeta-Wilson B, MacAvoy MG, Ding YS, Gelernter J, Carson RE. Amyloid-β burden and neuropsychological test performance in cognitively normal first-degree relatives at varying genetic risk for Alzheimer’s disease. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
Vandenberghe R, Thurfjell L, Van Laere K, Buckley CJ, Owenius R, Brooks DJ. Comparison between cerebellum and pons as reference regions for quantification of the amyloid imaging agents [18F]flutemetamol and [11C]PIB. Human Amyloid Imaging 2010 Meeting Abstracts. 2010 April 9. Abstract
This concludes a six-part series of the HAI Amyloid Imaging Conference. View a PDF of Part 6. See also Part 1, Part 2, Part 3, Part 4, Part 5. View PDF of the entire series.
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.