Let ’Er RIP: ALS/FTD Variant Plus Aging Unleash Deadly Kinase
Quick Links
Living up to its ominous acronym, RIPK1, the receptor-interacting serine/threonine-protein kinase 1 unleashes powerful signaling cascades that can fell cells within hours. Thankfully, several inhibitors normally keep these deadly doings in check. But what happens if this redundancy fails? In the August 23 Cell, scientists led by Junying Yuan of Harvard Medical School propose that when a genetic mutation hobbles an RIPK1 inhibitor called TBK1, and age quells another dubbed TAK1, ALS/FTD ensues. The theory helps explain how age exacerbates genetic susceptibility to disease, and may have implications beyond ALS/FTD, Yuan told Alzforum.
- TBK1 inhibits RIPK1 by phosphorylating the kinase.
- TAK1, another RIPK1 inhibitor, declines with age.
- Haploinsuffiency of each causes microgliosis, neurodegeneration, and ALS/FTD-like symptoms in mice.
Mutations in TBK1, a.k.a. TANK-binding kinase 1, account for more than 10 percent of familial cases of ALS/FTD (Feb 2015 news). The mutations dampen the protein’s expression, suggesting haploinsufficiency leads to disease (Freischmidt et al., 2017). Attempts to study animal models of TBK1-ALS/FTD have been hampered because animals that carry only a single copy of the gene appear normal, while TBK1 knockouts die before birth (Bonnard et al., 2000).
First author Daichao Xu and colleagues had a hunch that RIPK1, a kinase scientists have implicated in multiple neurodegenerative diseases, might somehow play a role in the death of the TBK1 knockouts (Jul 2017 news; Ito et al., 2016; and Sep 2017 news). Sure enough, they found that the mice were born healthy if one or both copies of the RIPK1 gene were replaced by a kinase-dead version. Comparing embryos carrying normal versus inactive RIPK1, they found that the kinase exacted the brunt of its toll in the fetal liver, where it triggered inflammation and apoptosis. Though there was no indication of neurodegeneration in the early brain, the data cast TBK1 as a RIPK1 inhibitor.
Through cell-culture, biochemical, and animal-model experiments, Xu and colleagues zeroed in on how TBK1 blocks RIPK1. They found it directly phosphorylated the protein at threonine-189, a residue that sits smack dab in the middle of the RIPK1 kinase domain. Interestingly, the only known substrate of RIPK1 is none other than RIPK1 itself: The protein forms dimers, which phosphorylate each other at serine-166, ultimately allowing the protein to hook up with other death-domain containing proteins and trigger apoptosis. TBK1 prevented this trans-phosphorylation, thus squelching RIPK1’s apoptotic activity.
Why, then, do TBK1-heterozygous mice develop normally, while people with a mutated copy of the gene develop ALS/FTD? The researchers suspected that aging exacerbates TBK1 deficiency. Having found that TBK1 blocks RIPK1, they wondered if another known RIPK1 inhibitor, TGFβ-activated kinase 1 (TAK1), takes up the slack when TBK1 falters (Geng et al., 2017). Indeed, they found that TAK1 was more active in mouse embryonic fibroblasts (MEFs) from TBK1 knockout mice than in fibroblasts from wild-type controls, suggesting that the kinase compensates for loss of TBK1.
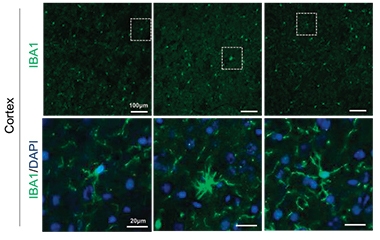
RIPK1 Awakens Microglia.
Microglia expanded and took on an activated, ameboid morphology in TBK1/TAK1 double heterozygous mice (middle), compared to wild-type (left) or mice expressing inactive RIPK1 (right). [Courtesy of Xu et al., Cell, 2018.]
Might TAK1 somehow lose its hold over RIPK1 during aging? To investigate this, the researchers sifted through gene-expression data from postmortem human brain samples and found 1.33-fold less TAK1 mRNA in people older than 60 than in people younger than 40. What’s more, prefrontal cortex samples from old donors had about 80 percent less of the protein than samples from young donors. Immunolabeling of brain sections suggested that microglia from aged donors expressed less TAK1 than microglia from young donors. In contrast, TBK1 expression held steady with age. Given that the average onset of ALS/FTD is around 60 years of age, the researchers proposed that age-related loss of TAK1 expression, in combination with a mutation in TBK1, might unfetter RIPK1 and lead to neurotoxicty.
The scientists put this idea to the test in an animal model, focusing on microglia. They generated TBK1-heterozygous mice in which myeloid cells were also heterozygous for TAK1. At six months of age, the mice had signs of microgliosis in the spinal cord and cortex, while TBK1-single-heterozygous mice did not. RNA sequencing of microglia in the double heterozygous mice revealed elevated expression of 40 genes involved in innate immunity, including cytokines and chemokines.
How would neurons fare amidst this inflammatory onslaught? The spinal cords of six-month-old, double-heterozygous animals had about 30 percent fewer motor axons, and abnormal myelination of existing ones, compared with mice heterozygous for TBK1 alone. Neuromuscular junctions denervated, and numerous neurons appeared to be in apoptotic demise. In the cortex, Xu found 15 percent fewer neurons in the double-heterozygous mice, while 6 percent of the remaining neurons had cytoplasmic inclusions of TDP-43—a hallmark of ALS/FTD. In both the cortex and spinal cord, RIPK1 activity was higher than in TBK1 heterozygotes.

Toxic TDP-43?
TBK1/TAK1 double-heterozygotes had neurons harboring cytoplasmic inclusions of TDP-43 (middle). Neurons from wild-type mice (left) or double-heterozygotes expressing an inactive form of RIPK1 (right) had none. [Courtesy of Xu et al., Cell 2018.]
The double-heterozygous animals had symptoms reminiscent of both ALS and FTD. At six months of age, the animals stopped standing on their hind legs, suggesting muscle weakness. The mice also appeared more anxious, as they hesitated more than other mice to explore an open field or unfamiliar sections of a maze.
None of these pathological or behavioral problems emerged in TBK1 single-heterozygous animals, nor in TBK1/TAK1 double-heterozygous mice expressing an inactive copy of RIPK1. In all, the findings suggest that halving the expression of both TBK1 and TAK1 in myeloid cells allows RIPK1 to trigger neuroinflammation, which trips off apoptosis in neurons.
Yuan proposed that TBK1 and TAK1 work together to block RIPK1-dependent inflammation and apoptosis. Though this could explain how ALS/FTD arises in people with a TBK1 mutation, Yuan speculated that TBK1 expression could be compromised in other situations. For example, TBK1 has anti-viral activity, so the microbes have evolved mechanisms to shut down its expression. Viral infections that occur in the context of aging might wear down defenses against RIPK1, she suggested. Viral expression has recently been linked to Alzheimer’s disease (Jun 2018 news). She added that waning TAK1 expression with age could also sensitize people to a range of neurodegenerative diseases beyond ALS/FTD, and she is studying what causes TAK1 expression to decline with age. Yuan previously reported that deficiency in optineurin, another ALS gene, promoted RIPK1 activity by stabilizing the kinase (Ito et al., 2016).
“This work provides additional support for the idea that mutations in TBK1 contribute to disease at least in part through modulation of RIPK1 activity in the aged brain,” commented Joseph Lewcock of Denali Therapeutics in South San Francisco. “When taken along with the data from optineurin-null animals previously published by the same group, these results suggest that RIPK1 inhibition may provide therapeutic benefit for at least a subset of ALS patients,” he wrote. Denali licensed a RIPK1 inhibitor program from Yuan and has begun Phase 1 testing in healthy volunteers.
Oleg Butovsky of Brigham and Women’s Hospital in Boston noted that while the study raises important questions, it is at odds with some previous reports. For one, microglia express very low levels of TAK1 and TBK1, according to data from Butovsky and others (Zhang et al., 2014, and publicly available database). He has compared the transcriptomes of microglia in wild-type and AD mice, and told Alzforum that microglia from neither expressed TAK1 (Sep 2017 news). The cells did express low levels of TBK1, which dropped by 1.5-fold in microglia that surrounded plaques. Au contraire, Yuan noted an RNA-Seq analysis of the cerebral cortex carried out at the late Ben Barres’s lab at Stanford University indicates that expression of TAK1, a.k.a. MAP3K7, is not low in microglia from people or from mice, and is comparable to expression of RIPK1, while TBK1 expression is even higher (Zhang et al., 2014).
Butovsky wondered whether infiltrating myeloid cells, rather than microglia, might play a role in the death cascade. A previous study led by Marco Prinz reported that knocking out TAK1 in microglia reduced inflammation and lessened axonal damage, a result seemingly at odds with Yuan’s current findings (Goldmann et al., 2013). Butovsky suggested that directly comparing the effects of limiting TAK1 deficiency to microglia, as opposed to all myeloid cells, might help clear up such discrepancies. —Jessica Shugart
References
News Citations
- TANK-Binding Kinase 1 Rumbles in as New ALS Gene
- Necroptosis Rampant in the Alzheimer’s Brain?
- Microglial Kinase Promotes DAM, Blocks Lysosomal Aβ Digestion
- Aberrant Networks in Alzheimer’s Tied to Herpes Viruses
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
Paper Citations
- Freischmidt A, Müller K, Ludolph AC, Weishaupt JH, Andersen PM. Association of Mutations in TBK1 With Sporadic and Familial Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. JAMA Neurol. 2017 Jan 1;74(1):110-113. PubMed.
- Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, Itié A, Wakeham A, Shahinian A, Henzel WJ, Elia AJ, Shillinglaw W, Mak TW, Cao Z, Yeh WC. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 2000 Sep 15;19(18):4976-85. PubMed.
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, Geng J, Amin P, DeWitt JP, Mookhtiar AK, Florez M, Ouchida AT, Fan JB, Pasparakis M, Kelliher MA, Ravits J, Yuan J. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016 Aug 5;353(6299):603-8. PubMed.
- Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, Mookhtiar AK, Zhao H, Xu D, Shan B, Najafov A, Gao G, Akira S, Yuan J. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun. 2017 Aug 25;8(1):359. PubMed.
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014 Sep 3;34(36):11929-47. PubMed.
- Goldmann T, Wieghofer P, Müller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, Luedde T, Heikenwalder M, Jung S, Prinz M. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci. 2013 Nov;16(11):1618-26. Epub 2013 Sep 29 PubMed.
External Citations
Further Reading
Primary Papers
- Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, Mifflin L, Pan L, Amin P, Li W, Shan B, Naito MG, Meng H, Li Y, Pan H, Aron L, Adiconis X, Levin JZ, Yankner BA, Yuan J. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell. 2018 Sep 6;174(6):1477-1491.e19. Epub 2018 Aug 23 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Denali Therapeutics
This manuscript from Xu et al. demonstrates that haploinsufficiency for TBK1, a gene known to contribute to the risk of ALS, is sufficient to lead to neurodegenerative phenotypes when on a background of mice lacking one copy of the kinase TAK1. Interestingly, these phenotypes are dependent on RIPK1 activity and are reversed in RIPK1 kinase dead mice. As aging results in a reduction of TAK1 expression in human brain, this work provides additional support for the idea that mutations in TBK1 contribute to disease at least in part through modulation of RIPK1 activity in the aged brain. When taken along with the data from optineurin (OPTN) null animals previously published by the same group, these results suggest that RIPK1 inhibition may provide therapeutic benefit for at least a subset of ALS patients.
Moving forward, it will be interesting to better understand whether the decrease in TAK1 expression observed in humans during aging has the same impact on RIPK1 signaling as what is observed in TAK1-heterozygous animals. In addition, gaining an improved understanding of the factors that drive RIPK1 activation in the presence of TBK1 or OPTN as well as finding clinically translatable biomarkers to measure RIPK1 activity will be important steps towards identifying a broader ALS patient population that may benefit from this approach, and may open the door to the application of RIPK1-based therapy in other chronic neurodegenerative indications.
Make a Comment
To make a comment you must login or register.