Necroptosis Rampant in the Alzheimer’s Brain?
Quick Links
Researchers have yet to solve the neuronal murder mystery in the Alzheimer’s brain, but one group believes they have found a smoking gun. Necrosomes—ugly blots that bespeak death by necroptosis—speckled postmortem brain tissue of people with Alzheimer’s disease, according to a study published July 24 in Nature Neuroscience. The researchers, led by Salvatore Oddo at Arizona State University in Tempe, reported that in people with AD, markers of this controlled form of cell death correlated with disease stage, memory slippage, tau pathology, and brain atrophy. In animal models of AD, ramping up necroptosis worsened cognition, while inhibiting it reduced neuronal loss. The researchers propose that disrupting this form of neuronal execution could be a therapeutic strategy.
“These findings suggest that necroptosis may play a direct role in cognitive dysfunction in people with AD,” commented Junying Yuan of Harvard Medical School in Boston.
Somewhere in between the programmed, clean death of apoptosis and the explosive, messy death of necrosis lies a middle path called necroptosis. First found to kill neurons following stroke, necroptosis is a highly regulated form of necrosis (see Jun 2005 news). It has since been found to play a hand in neurodegenerative diseases including amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS) (Feb 2014 news; Ito et al., 2016; Ofengeim et al., 2015).
Cell stress and inflammation trigger the pathway, whose cardinal feature is the formation of so-called “necrosomes.” These insoluble complexes of RIPK1 and RIPK3 kinases, together with the kinase MLKL, which functions as a hot poker of membranes, ultimately destroy the cell (reviewed in Grootjans et al., 2017).
To explore whether this mode of cell death ramps up in the AD brain, first author Antonella Caccamo and colleagues searched for markers of necroptosis in postmortem brain samples from 12 AD patients and 11 controls. They found elevated levels of RIPK1 and MLKL in insoluble fractions of AD brain. Immunofluorescence revealed RIPK1, RIPK3, and MLKL comingling, as well as more phosphorylated and dimerized MLKL, which is considered evidence of the protein’s executioner activity. More than half of phosphorylated MLKL resided in neurons, while about a third appeared in microglia and a tenth in astrocytes. The researchers also reported that around half of RIPK1 and MLKL co-localized with tau pathology.

Caught Red-Handed?
RIPK1 (red) and MLKL (blue) appear partnered in crime within neurons, though some colocalization appear in controls, in addition to elevated staining in AD. [Courtesy of Caccamo et al., Nature Neuroscience, 2017.]
To expand their findings to larger numbers of patients, the researchers compared microarray data on nearly 100 AD patients with controls. They again found elevated RIPK1 and MLKL, but not RIPK3, in people with AD. Similar associations appeared in a small gene expression data set from neurons obtained via laser capture. The researchers once again found that in people with AD, RIPK1, and MLKL, but not RIPK3, expression correlated with Braak stage and poor performance on the mini mental state exam (MMSE), while expression of RIPK1 alone correlated with brain atrophy. None of the necroptosis markers correlated with Aβ plaque load. The findings suggest that necroptosis correlates with tau pathology, neuronal death, and cognitive symptoms in AD, Oddo said. He was unable to explain why RIPK3 expression did not associate with various aspects of disease.
Besides its role in necroptosis, RIPK1 stimulates other signaling pathways related to inflammation and cellular stress. To understand more broadly how RIPK1 influenced gene-expression changes in the AD brain, the researchers generated a causal gene-interaction map. Using transcriptomic data taken from the postmortem ectorhinal cortices and anterior prefrontal cortices of controls or people who had died at different stages of AD, the researchers identified 819 genes whose expression covaried with RIPK1 expression. They leveraged relationships between certain genotypes (called cis-eQTLs) and RIPK1 expression to infer causality between RIPK1 expression and that of other genes. Notably, many of the genes influenced by RIPK1 were also differentially expressed in AD versus controls, including known AD risk genes. The findings raise the possibility that RIPK1 signaling orchestrates a chunk of the gene expression changes observed in the AD brain, the researchers proposed.
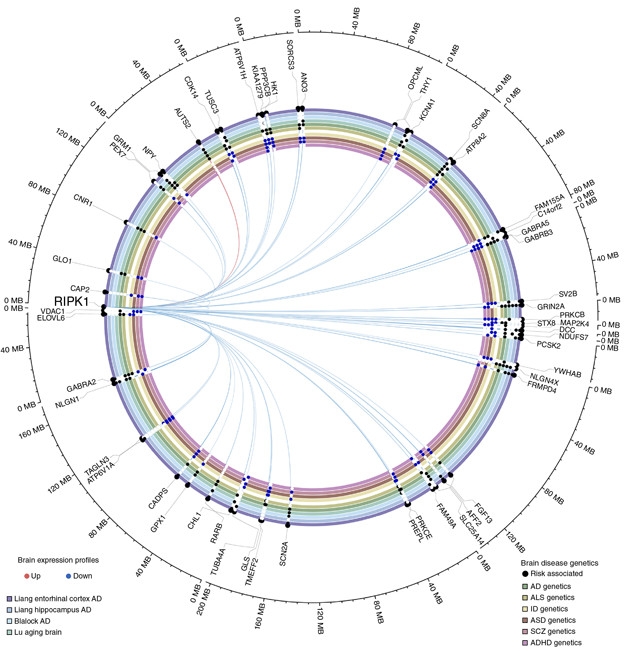
Master of Death? RIPK1 expression influences expression of myriad genes, including many that are differentially expressed in neurodegenerative disease compared to controls. [Courtesy of Caccamo et al., Nature Neuroscience, 2017.]
Finally, Caccamo and colleagues looked at the role of necroptosis in two AD mouse models: 5xFAD and APP/PS1 mice. Compared to non-transgenic mice, 5xFAD animals had soaring levels of RIPK1, MLKL, and phospho-MLKL proteins in their brains, in keeping with the substantial neuronal loss observed in that model. Treating 5xFAD mice with the necroptosis inhibitor Nec1s reduced the extent of their cell death. Behavioral analysis of these Nec1s-treated mice is only now ongoing, Oddo told Alzforum.
In contrast, APP/PS1 mice showed no signs of necroptosis, consistent with the lack of neuronal death observed in those animals. However, when the researchers transduced APP/PS1 animals with a constitutively active form of MLKL, thus switching on necroptosis, the animals performed worse on tests of spatial memory and suffered more neuronal loss than their untreated counterparts. Turning on necroptosis in the APP/PS1 animals had no effect on their Aβ load or levels of hyperphosphorylated tau, suggesting the pathway does not contribute to AD pathology. Aβ or tau pathology was not assessed in Nec1s-treated 5xFAD mice.
Oddo proposed a model in which necroptosis becomes activated, perhaps due to inflammation as well as other insults such as tau pathology. Cells dying in this manner then stimulate further inflammation, feeding a cycle of destruction. To Oddo, the next questions to address are what triggers necroptosis, as well as what roles neurons and glia play relative to each other in the death pathway. Oddo also plans to search for markers of necroptosis in postmortem brain tissue from people with other tauopathies, and to further characterize the death pathway in tau transgenic mice.
Oddo’s gene expression data place RIPK1 upstream of myriad gene expression changes in the AD brain, hinting that there could be more to the story than a simple death switch, commented Serge Przedborski of Columbia University in New York. He praised the neuron-specific gene-expression data as a useful addition to the field.
Przedborski, who previously reported that astrocytes from ALS patients trigger neuronal death via necroptosis in vitro (Feb 2014 news), cautioned that the poor quality, particularly the insufficient specificity, of antibodies for necrosome proteins have plagued the necroptosis field, rendering suspect the interpretation of immunofluorescence data. That said, Przedborski noted that regardless of whether it is bona fide necroptosis that drives cell death in the AD brain, animal experiments suggest that blocking the pathway could have therapeutic benefits through mechanisms that need further study. Yuan, who initially coined the term “necroptosis,” agreed that the pathway is an excellent target. “RIPK1 does all of the bad things downstream of TNFα,” she told Alzforum. She was also surprised to see necroptosis markers in neurons, as opposed to glial cells alone, and impressed by the correlation between Braak stage and necroptosis markers in Oddo’s study.
Shijun Zhang of Virginia Commonwealth University in Richmond, who has developed a small molecule that shields neurons from Aβ-induced damage and prevents necroptosis (Liu et al., 2015), commented that Oddo’s findings help link neuroinflammation—a phenomenon with strong genetic ties to AD risk—directly to neuronal loss.—Jessica Shugart
References
News Citations
- A New Program for Cell Death: Necroptosis Premiering in a Neuron Near You
- Death in a Dish: Astrocytes from ALS Patients Flick Necroptosis Switch in Motor Neurons
Research Models Citations
Paper Citations
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, Geng J, Amin P, DeWitt JP, Mookhtiar AK, Florez M, Ouchida AT, Fan JB, Pasparakis M, Kelliher MA, Ravits J, Yuan J. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016 Aug 5;353(6299):603-8. PubMed.
- Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, Geng J, Py B, Zhou W, Amin P, Berlink Lima J, Qi C, Yu Q, Trapp B, Yuan J. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015 Mar 24;10(11):1836-49. PubMed.
- Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017 Jul;24(7):1184-1195. Epub 2017 May 12 PubMed.
- Liu K, Chojnacki JE, Wade EE, Saathoff JM, Lesnefsky EJ, Chen Q, Zhang S. Bivalent Compound 17MN Exerts Neuroprotection through Interaction at Multiple Sites in a Cellular Model of Alzheimer's Disease. J Alzheimers Dis. 2015;47(4):1021-33. PubMed.
Further Reading
Primary Papers
- Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, Belfiore R, Winslow W, Oddo S. Necroptosis activation in Alzheimer's disease. Nat Neurosci. 2017 Sep;20(9):1236-1246. Epub 2017 Jul 24 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.