Brain Biopsies and FinnGen Form Wellspring for Functional Genomics
Quick Links
Tucked into the waterways of a Nordic landscape, moose roaming its woods near the Russian border, Kuopio, population 300,000, is both small and remote. Yet this Finnish city lies in the center of a new approach to functional genomics and personalized medicine research on the global scourge of age-related neurodegeneration. Its secret? Close integration between neurosurgeons and all manner of biologists, plus data collected across national health registries, tissue banks, and genotyping cohorts.
- In Finland, extensive biobanking and genotyping provide human tissue for ADRD research.
- Combined with mouse models, this system empowers functional genomics research.
- Currently under scrutiny: ABI3, APP, C9ORF72, GRN, PLCG2, TMEM106B, and other genes.
In this technologically advanced nation of 5.5 million people, Mikko Hiltunen directs the University of Eastern Finland's overarching Neuroscience Research Community, UEF NEURO-RC. Among its principal investigators, a select handful collaborate on a range of functional studies that draw on Kuopio's unique practice of extensive biopsy collection and tissue banking. These, in turn, draw on routine brain surgeries that treat age-related idiopathic hydrocephalus (iNPH). UEF NEURO-RC's system of interlocking biopsy and basic research enables multilevel exploration of research questions across tissue types and through multiple levels of analysis, from electrophysiology of brain slices to transcriptomic, proteomic, and biomarker research of brain parenchyma and fluid samples. (For more on this system, see Parts 1, 2, 3, and 4 of this series.)
ADRD genetics boasts a growing list of GWAS hits. The painstaking work of figuring out how each of these operate in cells challenges scientists at UEFs Neuro-RC and around the world. Hiltunen's group has coupled Kuopio's hydrocephalus biopsy banking protocol and its attendant phenotypic data to a translational and mouse research program he calls Personalized Medicine for Microglial-associated genetic variants in Alzheimer's Disease. Aka PMG-AD, the program stands on three legs: the iNPH sampling protocol, FinnGen/Finnish biobanks, and mouse models. PMG-AD was established in collaboration with the German Center for Neurodegenerative Diseases, University of Lille, France, University of Helsinki, and Turku PET Centre, Finland.

PMG-AD. This research program taps cohort studies for carriers of genetic variants of interest, and invites them into longitudinal imaging studies that use their blood for cell-based and multiomic analyses (top). Mouse lines of those variants add a model for deeper mechanistic studies (bottom). [Courtesy of M. Hiltunen.]
At its core, PMG-AD takes discoveries from human genetics, for example the protective A673T "Icelandic" APP or PLCG2 Pro522Arg variants, finds carriers among Finnish population and biobank cohorts, and asks them to join detailed longitudinal aging studies. Likewise, carriers of AD and FTD risk variants, e.g. ABI3Ser209Phe or GRN rs5848, get invited to come to UEF or the University of Helsinki for research monitoring their prodromal stages. Ditto for carriers of the C9ORF72 hexanucleotide expansion, or TREM2 or TYROBP mutations.
Hiltunen's long-term goal is to gather rare variant carriers within Finnish biobanks into a cohort study that characterizes their preclinical pathophysiology with all major tools available, probing biomolecules, cells, tissues, and the brain. The idea draws inspiration from what the Dominantly Inherited Alzheimer's Network (DIAN) has pioneered for autosomal-dominant AD. Fortuitously for Hiltunen's team, carriers of all the above gene variants showed up not only in Finnish population cohorts but also in the hydrocephalus cohort led by neurosurgeon Ville Leinonen of Kuopio University Hospital. This means their shunt biopsies supply brain tissue and fluids for PMG-AD research projects (see Parts 1 and 2).
Can researchers pull this off in real life? It appears so, according to a pilot FinnGen recall study, albeit not yet with preclinical variant carriers, but with people whose records suggest they have early AD (Julkunen et al., 2023). Also helpful here is Finland's 2012 Biobank Law, which enables participants to donate samples with a broad consent to medical research.
A “younger sibling” to the better-known U.K. Biobank, FinnGen started in 2017. This pre-competitive partnership unites nine Finnish biobanks and five Finnish university hospitals into a hub co-funded by the Finnish government and 13 international pharma companies. Led by Aarno Palotie of the University of Helsinki and Massachusetts General Hospital, the project's stated goal is to include 500,000 Finns, nearly 10 percent of the country's population, by the end of 2023. For phenotype data, FinnGen takes digital records from hospital visits, drug purchases/reimbursement, and from longitudinal health register data collected for every resident of this Nordic nation since 1969. For genotype data, FinnGen uses legacy or newly collected blood samples and runs them on a Thermo Fisher custom array tweaked toward the Finnish gene pool.

Banks Everywhere. Nine Finnish Biobanks, seven regional ones, and three nationwide ones (gray circles) supply FinnGen with samples. [Courtesy of Nature.]
Genetically speaking, FinnGen's population is more homogeneous than the U.K.'s, because Finland lacks the population admixture arising from the U.K.’s colonial history. This makes rare, high-impact disease variants easier to find in the Finnish population.
This is not the only difference. Starting in 2006, the U.K. Biobank sought participants by mail and enrolled them with structured, in-person interviews. Hence it has detailed, standardized phenotypic and behavioral data gathered at those visits, but suffers from "healthy volunteer bias." In contrast, FinnGen pulls samples from the country's extensive network of biobanks and taps digital records from hospital visits and research studies. This makes FinnGen stacked with people who have seen specialists or been in hospitals (Rodosthenous et al., 2022). For example, a published analysis of the first 224,737 participants, median age 73, put FinnGen's Alzheimer's disease prevalence at 2.7 percent compared to 0.2 percent in the U.K. Biobank (Kurki et al., 2023). This number is based on data release 5, which is public. Data freeze 11, accessible to member scientists, already shows three times as many AD cases, Hiltunen told Alzforum.
FinnGen works by give and take. Hiltunen contributed DNA on 2,000 AD patients, plus age-matched controls, whom his genetics group has been studying over the past 20 years. Likewise, Leinonen contributed data and samples of 1,000 hydrocephalus patients. Importantly though, Hiltunen also pulls information from FinnGen to drive his lab's PMG-AD project. FinnGen is where scientists can access data and samples from individual Finns with gene variants of interest. As Hiltunen monitors the growing FinnGen data, he frequently finds, and tests, carriers of neurodegenerative variants that are being newly discovered in other genetic studies, such as the European Alzheimer and Dementia Biobank (EADB). These are some of the people he hopes to invite to join research before their disease manifests itself—or to learn how their variant keeps them cognitively well into old age.
The third leg of this PMG-AD approach is work with mouse models. Scientists led by UEF's Mari Takalo subject mice and their tissues to the same experiments she runs on samples from biobanks and iNPH variant carriers. These days, she focuses on the topic that preoccupies ADRD scientists around the world, that is, the nexus between the innate immune system, glia, and neurons during the “cellular phase” of early AD. "Whichever variant we are studying, we are characterizing if it influences key microglial functions such as phagocytosis, inflammatory response, lipid metabolism, or viability of the microglia," Takalo told Alzforum.
Her team's methods range broadly from PET imaging to cell-based assays derived from carrier monocytes, iPSCs, or organoids, and to multiomic analyses. In cases where the stars align and both FinnGen and the iNPH cohort harbor carriers of a variant of interest, Kuopio scientists have access to an unparalleled collection of tissues: frontal cortex and dura, intraventricular and lumbar CSF, cells thereof, blood and skin-derived cells including monocytes, fibroblasts to be used to generate iPSCs and organoids. All this from a living, aging carrier who is being followed cognitively, with neuroimaging and further fluid sampling as needed for their care.
This armamentarium enables the scientists to conduct frequent cross-checks to ensure that a new finding in mice is true in humans, i.e., is relevant to disease, or that a finding in humans can be experimentally explored in greater depth in mice. Hiltunen hopes this approach will fill in the sought-after pathways that lead from genes to AD, CAA, or ALS/FTD.
To illustrate how this concept works in the lab, this story summarizes four example projects: on APP, PLCg2, progranulin, and C9ORF72. Similar work is happening with Abi33, TREM2, and TYROBP. Work is ongoing, or at early stages.
APP
First described in 2012, the APP A673T variant protects against AD (Jonsson et al., 2012). Naming it the "Icelandic mutation" was perhaps a tad hasty, as it occurs in about 30,000 lucky Finns. Six years ago, when Hiltunen and colleagues submitted a paper showing that A673T carriers had 30 percent less Aβ42 in their plasma, reviewers wanted to know if that was true in their brains and CSF, as well. "We were unable to answer that because we had no such tissue in our original genetic sample," Hiltunen recalled (Martiskainen et al., 2017).
Then three APP A673T carriers cropped up in the iNPH biopsy cohort. Lo and behold, their CSF was indeed low in Aβ42. "Typically in AD, CSF Aβ42 reduction is taken to mean amyloid deposition in the brain," Hiltunen teased. But no, their brain biopsies, taken during shunt surgery (see Part 2), showed the cortices of these aging carriers to be free of amyloid pathology. In their CSF, sAPPβ, the product of BACE cleavage, was lower, while sAPPα, the product of α-secretase cleavage, was higher than in well-matched noncarriers.

Like Blood Like Brain. sAPPβ and Aβ42 levels are lower, sAPPα is higher, in CSF samples of APP A673T variant carriers (light bars) than controls (dark bars). [Courtesy of Wittrahm et al., 2023.]
Also consider results of mass-spec-based proteomic, phospho-proteomic, and RNA analyses of CSF and frontal cortex in the iNPH biopsy cohort. Rebekka Wittrahm, Takalo, and colleagues in the lab matched the cohort's three APP A673T carriers with three noncarriers of the same APOE genotype, sex, and age. They added Braak-staged AD postmortem tissue for another comparison. They found that specific pathways that were upregulated in the APP A673T carriers were downregulated in their fellow noncarriers and even more so in postmortem AD brains of increasing disease stage. "We are seeing the same pathways changed in opposite ways in living APP Icelandic variant carriers versus pathological Alzheimer's cases," Hiltunen said.
Notably, subunits of protein phosphatases that dephosphorylate tau were at several-fold higher levels in the cortices of protective variant carriers than in AD cases, possibly tying non-amyloidogenic APP cleavage to tau modification. The variant's impact appears neuronal, as microglial phagocytic capacity and maintenance seemed unchanged in APP A673T carriers. Ditto for their astrocytes; the study has not looked at oligodendrocytes, though. (For details, see Wittrahm et al. 2023).
In cell-based experiments pitting the protective Icelandic variant against both the pathogenic Swedish and London APP variants, A673T proved “stronger.” It overpowered both these mutations by lowering sAPPβ and raising sAPPα. "Wherever you look, this mutation has beneficial effects, and we confirmed that it operates through APP cleavage," Hiltunen said.
These data would appear to support BACE inhibition as a therapeutic approach. Pharma companies have not revived work on their BACE inhibitor drugs, which scientists agree could work safely at low doses that aim for no more than 30 percent Aβ reduction—in the range Wittrahm reported to be the case in APP A673T CSF.
Following his 2017 report of Aβ42 reduction in the blood of such carriers, Hiltunen received pharma requests to measure Aβ42 in the CSF of FinnGen participants carrying APP A673T. He was unable to do so at the time because FinnGen does not collect CSF, and requesting a lumbar puncture from healthy people for research is not permitted in Finland. So, it was the iNPH cohort that opened a window into the APP A673T brain and answered the old question about its effect in CSF. As FinnGen is nearing its first goal of screening and genotyping 500,000 people, the Kuopio scientists are hoping that CSF sampling can be added in its next phase. "This will help us get endophenotype data so that we can connect participants' genetics to their phenotype," Hiltunen told Alzforum. Meanwhile, a deeper analysis of the APP A673T carriers' cognitive preservation as they age is ongoing, Hiltunen said.
PLCγ2
In the current iNPH cohort, two shunt recipients carry a rare variant that fascinates scientists because it protects against AD, FTD, and Lewy body disease. Unlike APP A673T, this one works through microglia, not neurons. It is the P522R coding variant of PLCG2, which encodes the membrane-associated phospholipase C gamma 2 (PLCγ2). In microglia, PLCγ2 acts downstream of TREM2 in that receptor's signaling pathway. Brain from these two iNPH shunt recipients show no sign of amyloid pathology, Takalo told Alzforum; their tissues are banked and awaiting deeper analyses.

Microglial Pathway. Signal transduction downstream of the microglial TREM2 receptor is drawing intense scrutiny these days. The Kuopio team is studying DAP12 and PLCγ2 variants found in FinnGen and iNPH cohorts. [Courtesy of M. Hiltunen.]
In their mechanistic exploration of the PLCγ2-P522R variant, the Kuopio scientists have previously reported that it activates microglia in mice, as per gene expression, functional assays, and TSPO PET (Takalo et al, 2020). Since then, their respect for its prowess has only grown. It turns out that among its carriers in the Finnish FINGERS cohort are several asymptomatic individuals, who are cognitively normal in their 80s despite also having two APOE4 alleles.
How could PLCγ2-P522R possibly override the effect of ApoE4? For one, the scientists are finding that the lipid droplets that form in human PLCγ2-P522R microglial cultures stressed with LPS or myelin are smaller. It appears they do not fuse with larger lipid droplets. For another, PLCγ2-P522R transgenic mice express more of certain fatty-acid-binding and cholesterol-metabolizing genes. In essence, they appear better equipped to eliminate waste products as they age.
Feeding this interest in lipid metabolism is a related PMG-AD project with iNPH/FinnGen participants. It looks at pathogenic variants in TYROBP. This gene encodes the Dap12 receptor wedged between Trem2 and PLCγ2 in the same microglial signaling pathway. Using biopsy tissues, the scientists are currently extending a prior study (Natunen et al., 2020). It reported low microglial clustering around plaques in Type 2 diabetes induced in AD mouse models, and in amyloid plaques of iNPH patients with diabetes. Dap12 and PI3K-Akt appear to be involved. PI3-Akt also showed up in Takalo's lipid droplet study of PLCγ2-P522R cells.
For mechanistic studies of how PLCγ2-P522R operates in the presence of amyloid, these mice have been crossed to APP/PS1 delta E9 transgenic mice; analyses are ongoing. Will their plaques be denser, dystrophic neurites fewer, synaptic pathology milder? Early hints are that the crosses do have less amyloid. It looks as if PLCγ2-P522R microglia more actively surveil and compact plaques, but most types of tissue are still being analyzed, Takalo said.
How can scientists study people with a rare, protective variant? It's a conundrum because, by definition, there are few of them, and they don't have the disease in question. Once again, Hiltunen is looking to FinnGen. One question he wants to ask: Might the B cells of PLCγ2-P522R be mounting a better antibody response to the presence of accumulating Aβ? After all, peripheral immune cells express PLCγ2, as well. B lymphocytes make it as part of their maturation while fighting an immune challenge. "We assume that while the B cells are more activated in these carriers, they produce a factor that is beneficial in the periphery, then enters the brain," Hiltunen said. A separate aging cohort study, led by Henne Holstege at Vrije Universiteit Amsterdam, also detected a slightly more responsive immune system, including more sensitive B cells, in PLCγ2-P522R carriers (Diks et al., 2023).
Progranulin
The GRN gene popped up as genome-wide significant in an EADB GWAS FinnGen cohort (Bellenguez et al., 2022). Its rs5848 variant increases risk of ADRD. Fourteen years ago, Hiltunen's group had reported that rs5848 brings down the age of AD onset in men—to great skepticism, as he recalls (Viswanathan et al., 2009; Kämäläinen et al., 2012).
Luckily, the GRN rs5848 variant occurs among Leinonen's iNPH shunt patients. Progranulin protein levels wane in men with age, and the rs5848 variant decreases blood levels by another 25 percent in those homozygous for the risk allele. This puts them at risk of neurodegeneration, though this protein reduction is a subtler phenotype than a loss-of-function mutation or a mouse knockout.
In brain biopsy tissue of 60- to 75-year-old men with iNPH who were homozygous for the 5848 TT risk allele, Heli Jeskanen in Hiltunen's lab detected a trend toward lower progranulin protein levels than in controls. She also noticed that the number of microglia was the same as in controls but, curiously, more of them seemed to be heading toward amyloid plaques, which appeared smaller, more rounded. Studying microglia-like cells the scientists derive from these same patients' monocytes, Jeskanen found that rs5848 carrier cells took up more myelin and got more riled up by LPS. Something about them seemed more reactive. These data replicate, in human cortex and blood cells, a progranulin knockout/APP transgenic study that had reported hyperactive microglia (Goetzl et al., 2019). "This is translation from mouse to human," Hiltunen said. More work is ongoing.
Another variant of intense interest that occurs in both FinnGen and the Kuopio iNPH cohort is TMEM106B rs13237518 (Katsumata et al., 2022). Hiltunen suspects TMEM106B and progranulin act in the same pathway, and his team is dissecting the mechanisms of both genes using the brain biopsies, CSF, and plasma samples from the iNPH patients. The cortical biopsies are being explored functionally in Tarja Malm's electrophysiology group (see Part 3); the CSF and plasma from carriers are studied in collaboration with Henrik Zetterberg and colleagues in Gothenberg, to look for changes in proteome and in markers of inflammation and synaptic damage.
"We bank these tissues, hence have access to additional material for other studies. Whatever comes up in CSF, we can check whether it also happens in cortex, with gene expression or function there, or whether it correlates with something we see in plasma," Hiltunen said.
Last But Not Least: C9ORF72
iNPH biopsies can pry open the door to identifying people with structural mutations—expansions, inversions, or deletions—that GWAS cannot pick up. Consider the C9ORF72 hexanucleotide expansion. It is the most common cause of FTD in Finland. But how can scientists find its carriers before they show symptoms? Some carriers are likely in FinnGen. Alas, FinnGen determines only SNPs. The elaborate genetic methods needed to detect the C9ORF72 expansion can't be applied to this many people. "We cannot see C9 expansions in FinnGen," Hiltunen said.
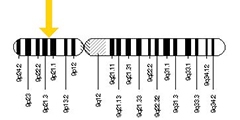
Find a Needle in the Haystack. Kuopio’s iNPH cohort enabled a genetic search strategy to pick out likely C9ORF72 hexanucleotide expansion carriers in FinnGen.
As it happened, the Kuopio iNPH cohort contains eight carriers (Korhonen et al., 2019). A genetic bootstrapping hunt ensued. Leinonen shared DNA and data from these carriers with the EADB. The world's largest AD genomic consortium, EADB continues to surface new disease variants (e.g. Nov 2022 news). Knowing which iNPH shunt patients had the C9ORF72 expansion enabled Hannah Rostalski in Annakaisa Haapasalo's lab at UEF to determine which GWAS proxy markers in EADB are associated with this expansion. Those proxies helped her define co-inherited SNP combinations, aka haplotype blocks, that can flag any person as likely carrying a C9ORF expansion. Once plugged into FinnGen, which at the time of this hunt had genotype data on 218,792 Finns, these haplotypes pulled out candidates. When the scientists checked these candidates' clinical diagnoses, lo and behold, they had FTLD and/or ALS (Rostalski et al., 2021).
Haapasalo and colleagues are now inviting these FinnGen participants, both those with a diagnosis and asymptomatic expansion carriers, into a research study to formally confirm the expansion in them with clinical-grade testing, and then to study their progression as part of UEF's translational medicine program.—Gabrielle Strobel
References
News Citations
- Fresh Brain Every Friday: Biopsies Transform Alzheimer's Science
- A Day's Work: Cortex Biopsy Comes Out. Shunt Goes In. Patient Goes Home.
- Brain Tissue From Living People with Amyloid Plaques Can Fire in a Dish
- Cortical Biopsies Hint at Start of Alzheimer's 'Cellular Phase'
- Rare Variants in Lipid Transporter Genes Increase Risk for Alzheimer’s Disease
Mutations Citations
Research Models Citations
Paper Citations
- Julkunen V, Schwarz C, Kalapudas J, Hallikainen M, Piironen A-K, Mannermaa A, Kujala H, Laitinen T, Kosma V-M, Paajanen T, Kalviainen R, Hiltunen M, Herukka S-K, Karkkainen S, Kokkola T, Urjansson M, FinnGen, Perola M, Palotie A, Vuoksimaa E, Runz H. A FinnGen pilot clinical recall study for Alzheimer's disease. 2023 Feb 08 10.1101/2023.02.06.23285534 (version 1) medRxiv.
- Rodosthenous RS, Niemi ME, Kallio L, Perala M, Terho P, Knopp T, Punkka E, Makkonen EM, Nurmi P, Makela J, Wihuri P, Hautalahti M, Moffatt C, Martini P, Germine L, Makela VA, Karhunen OA, Lahti J, Hiekkalinna TS, Jyrhama T, Shen HY, Runz H, Palotie A, Perola M, Ganna A, FinnGen. Recontacting biobank participants to collect lifestyle, behavioural and cognitive information via online questionnaires: lessons from a pilot study within FinnGen. BMJ Open. 2022 Oct 5;12(10):e064695. PubMed.
- Kurki MI, Karjalainen J, Palta P, Sipilä TP, Kristiansson K, Donner KM, Reeve MP, Laivuori H, Aavikko M, Kaunisto MA, Loukola A, Lahtela E, Mattsson H, Laiho P, Della Briotta Parolo P, Lehisto AA, Kanai M, Mars N, Rämö J, Kiiskinen T, Heyne HO, Veerapen K, Rüeger S, Lemmelä S, Zhou W, Ruotsalainen S, Pärn K, Hiekkalinna T, Koskelainen S, Paajanen T, Llorens V, Gracia-Tabuenca J, Siirtola H, Reis K, Elnahas AG, Sun B, Foley CN, Aalto-Setälä K, Alasoo K, Arvas M, Auro K, Biswas S, Bizaki-Vallaskangas A, Carpen O, Chen CY, Dada OA, Ding Z, Ehm MG, Eklund K, Färkkilä M, Finucane H, Ganna A, Ghazal A, Graham RR, Green EM, Hakanen A, Hautalahti M, Hedman ÅK, Hiltunen M, Hinttala R, Hovatta I, Hu X, Huertas-Vazquez A, Huilaja L, Hunkapiller J, Jacob H, Jensen JN, Joensuu H, John S, Julkunen V, Jung M, Junttila J, Kaarniranta K, Kähönen M, Kajanne R, Kallio L, Kälviäinen R, Kaprio J, FinnGen, Kerimov N, Kettunen J, Kilpeläinen E, Kilpi T, Klinger K, Kosma VM, Kuopio T, Kurra V, Laisk T, Laukkanen J, Lawless N, Liu A, Longerich S, Mägi R, Mäkelä J, Mäkitie A, Malarstig A, Mannermaa A, Maranville J, Matakidou A, Meretoja T, Mozaffari SV, Niemi ME, Niemi M, Niiranen T, O Donnell CJ, Obeidat ME, Okafo G, Ollila HM, Palomäki A, Palotie T, Partanen J, Paul DS, Pelkonen M, Pendergrass RK, Petrovski S, Pitkäranta A, Platt A, Pulford D, Punkka E, Pussinen P, Raghavan N, Rahimov F, Rajpal D, Renaud NA, Riley-Gillis B, Rodosthenous R, Saarentaus E, Salminen A, Salminen E, Salomaa V, Schleutker J, Serpi R, Shen HY, Siegel R, Silander K, Siltanen S, Soini S, Soininen H, Sul JH, Tachmazidou I, Tasanen K, Tienari P, Toppila-Salmi S, Tukiainen T, Tuomi T, Turunen JA, Ulirsch JC, Vaura F, Virolainen P, Waring J, Waterworth D, Yang R, Nelis M, Reigo A, Metspalu A, Milani L, Esko T, Fox C, Havulinna AS, Perola M, Ripatti S, Jalanko A, Laitinen T, Mäkelä TP, Plenge R, McCarthy M, Runz H, Daly MJ, Palotie A. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023 Jan;613(7944):508-518. Epub 2023 Jan 18 PubMed.
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012 Aug 2;488(7409):96-9. PubMed.
- Martiskainen H, Herukka SK, Stančáková A, Paananen J, Soininen H, Kuusisto J, Laakso M, Hiltunen M. Decreased plasma β-amyloid in the Alzheimer's disease APP A673T variant carriers. Ann Neurol. 2017 Jul;82(1):128-132. PubMed.
- Wittrahm R, Takalo M, Kuulasmaa T, Mäkinen PM, Mäkinen P, Končarević S, Fartzdinov V, Selzer S, Kokkola T, Antikainen L, Martiskainen H, Kemppainen S, Marttinen M, Jeskanen H, Rostalski H, Rahunen E, Kivipelto M, Ngandu T, Natunen T, Lambert JC, Tanzi RE, Kim DY, Rauramaa T, Herukka SK, Soininen H, Laakso M, Pike I, Leinonen V, Haapasalo A, Hiltunen M. Protective Alzheimer's disease-associated APP A673T variant predominantly decreases sAPPβ levels in cerebrospinal fluid and 2D/3D cell culture models. Neurobiol Dis. 2023 Jun 15;182:106140. Epub 2023 Apr 28 PubMed.
- Takalo M, Wittrahm R, Wefers B, Parhizkar S, Jokivarsi K, Kuulasmaa T, Mäkinen P, Martiskainen H, Wurst W, Xiang X, Marttinen M, Poutiainen P, Haapasalo A, Hiltunen M, Haass C. The Alzheimer's disease-associated protective Plcγ2-P522R variant promotes immune functions. Mol Neurodegener. 2020 Sep 11;15(1):52. PubMed.
- Natunen T, Martiskainen H, Marttinen M, Gabbouj S, Koivisto H, Kemppainen S, Kaipainen S, Takalo M, Svobodová H, Leppänen L, Kemiläinen B, Ryhänen S, Kuulasmaa T, Rahunen E, Juutinen S, Mäkinen P, Miettinen P, Rauramaa T, Pihlajamäki J, Haapasalo A, Leinonen V, Tanila H, Hiltunen M. Diabetic phenotype in mouse and humans reduces the number of microglia around β-amyloid plaques. Mol Neurodegener. 2020 Nov 10;15(1):66. PubMed.
- Diks AM, Teodosio C, de Mooij B, Groenland RJ, Naber BA, de Laat IF, Vloemans SA, Rohde S, de Jonge MI, Lorenz L, Horsten D, van Dongen JJ, Berkowska MA, Holstege H. Carriers of the p.P522R variant in PLCγ2 have a slightly more responsive immune system. Mol Neurodegener. 2023 Apr 20;18(1):25. PubMed.
- Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, Holmans PA, Boland A, Damotte V, van der Lee SJ, Costa MR, Kuulasmaa T, Yang Q, de Rojas I, Bis JC, Yaqub A, Prokic I, Chapuis J, Ahmad S, Giedraitis V, Aarsland D, Garcia-Gonzalez P, Abdelnour C, Alarcón-Martín E, Alcolea D, Alegret M, Alvarez I, Álvarez V, Armstrong NJ, Tsolaki A, Antúnez C, Appollonio I, Arcaro M, Archetti S, Pastor AA, Arosio B, Athanasiu L, Bailly H, Banaj N, Baquero M, Barral S, Beiser A, Pastor AB, Below JE, Benchek P, Benussi L, Berr C, Besse C, Bessi V, Binetti G, Bizarro A, Blesa R, Boada M, Boerwinkle E, Borroni B, Boschi S, Bossù P, Bråthen G, Bressler J, Bresner C, Brodaty H, Brookes KJ, Brusco LI, Buiza-Rueda D, Bûrger K, Burholt V, Bush WS, Calero M, Cantwell LB, Chene G, Chung J, Cuccaro ML, Carracedo Á, Cecchetti R, Cervera-Carles L, Charbonnier C, Chen HH, Chillotti C, Ciccone S, Claassen JA, Clark C, Conti E, Corma-Gómez A, Costantini E, Custodero C, Daian D, Dalmasso MC, Daniele A, Dardiotis E, Dartigues JF, de Deyn PP, de Paiva Lopes K, de Witte LD, Debette S, Deckert J, Del Ser T, Denning N, DeStefano A, Dichgans M, Diehl-Schmid J, Diez-Fairen M, Rossi PD, Djurovic S, Duron E, Düzel E, Dufouil C, Eiriksdottir G, Engelborghs S, Escott-Price V, Espinosa A, Ewers M, Faber KM, Fabrizio T, Nielsen SF, Fardo DW, Farotti L, Fenoglio C, Fernández-Fuertes M, Ferrari R, Ferreira CB, Ferri E, Fin B, Fischer P, Fladby T, Fließbach K, Fongang B, Fornage M, Fortea J, Foroud TM, Fostinelli S, Fox NC, Franco-Macías E, Bullido MJ, Frank-García A, Froelich L, Fulton-Howard B, Galimberti D, García-Alberca JM, García-González P, Garcia-Madrona S, Garcia-Ribas G, Ghidoni R, Giegling I, Giorgio G, Goate AM, Goldhardt O, Gomez-Fonseca D, González-Pérez A, Graff C, Grande G, Green E, Grimmer T, Grünblatt E, Grunin M, Gudnason V, Guetta-Baranes T, Haapasalo A, Hadjigeorgiou G, Haines JL, Hamilton-Nelson KL, Hampel H, Hanon O, Hardy J, Hartmann AM, Hausner L, Harwood J, Heilmann-Heimbach S, Helisalmi S, Heneka MT, Hernández I, Herrmann MJ, Hoffmann P, Holmes C, Holstege H, Vilas RH, Hulsman M, Humphrey J, Biessels GJ, Jian X, Johansson C, Jun GR, Kastumata Y, Kauwe J, Kehoe PG, Kilander L, Ståhlbom AK, Kivipelto M, Koivisto A, Kornhuber J, Kosmidis MH, Kukull WA, Kuksa PP, Kunkle BW, Kuzma AB, Lage C, Laukka EJ, Launer L, Lauria A, Lee CY, Lehtisalo J, Lerch O, Lleó A, Longstreth W Jr, Lopez O, de Munain AL, Love S, Löwemark M, Luckcuck L, Lunetta KL, Ma Y, Macías J, MacLeod CA, Maier W, Mangialasche F, Spallazzi M, Marquié M, Marshall R, Martin ER, Montes AM, Rodríguez CM, Masullo C, Mayeux R, Mead S, Mecocci P, Medina M, Meggy A, Mehrabian S, Mendoza S, Menéndez-González M, Mir P, Moebus S, Mol M, Molina-Porcel L, Montrreal L, Morelli L, Moreno F, Morgan K, Mosley T, Nöthen MM, Muchnik C, Mukherjee S, Nacmias B, Ngandu T, Nicolas G, Nordestgaard BG, Olaso R, Orellana A, Orsini M, Ortega G, Padovani A, Paolo C, Papenberg G, Parnetti L, Pasquier F, Pastor P, Peloso G, Pérez-Cordón A, Pérez-Tur J, Pericard P, Peters O, Pijnenburg YA, Pineda JA, Piñol-Ripoll G, Pisanu C, Polak T, Popp J, Posthuma D, Priller J, Puerta R, Quenez O, Quintela I, Thomassen JQ, Rábano A, Rainero I, Rajabli F, Ramakers I, Real LM, Reinders MJ, Reitz C, Reyes-Dumeyer D, Ridge P, Riedel-Heller S, Riederer P, Roberto N, Rodriguez-Rodriguez E, Rongve A, Allende IR, Rosende-Roca M, Royo JL, Rubino E, Rujescu D, Sáez ME, Sakka P, Saltvedt I, Sanabria Á, Sánchez-Arjona MB, Sanchez-Garcia F, Juan PS, Sánchez-Valle R, Sando SB, Sarnowski C, Satizabal CL, Scamosci M, Scarmeas N, Scarpini E, Scheltens P, Scherbaum N, Scherer M, Schmid M, Schneider A, Schott JM, Selbæk G, Seripa D, Serrano M, Sha J, Shadrin AA, Skrobot O, Slifer S, Snijders GJ, Soininen H, Solfrizzi V, Solomon A, Song Y, Sorbi S, Sotolongo-Grau O, Spalletta G, Spottke A, Squassina A, Stordal E, Tartan JP, Tárraga L, Tesí N, Thalamuthu A, Thomas T, Tosto G, Traykov L, Tremolizzo L, Tybjærg-Hansen A, Uitterlinden A, Ullgren A, Ulstein I, Valero S, Valladares O, Broeckhoven CV, Vance J, Vardarajan BN, van der Lugt A, Dongen JV, van Rooij J, van Swieten J, Vandenberghe R, Verhey F, Vidal JS, Vogelgsang J, Vyhnalek M, Wagner M, Wallon D, Wang LS, Wang R, Weinhold L, Wiltfang J, Windle G, Woods B, Yannakoulia M, Zare H, Zhao Y, Zhang X, Zhu C, Zulaica M, EADB, GR@ACE, DEGESCO, EADI, GERAD, Demgene, FinnGen, ADGC, CHARGE, Farrer LA, Psaty BM, Ghanbari M, Raj T, Sachdev P, Mather K, Jessen F, Ikram MA, de Mendonça A, Hort J, Tsolaki M, Pericak-Vance MA, Amouyel P, Williams J, Frikke-Schmidt R, Clarimon J, Deleuze JF, Rossi G, Seshadri S, Andreassen OA, Ingelsson M, Hiltunen M, Sleegers K, Schellenberg GD, van Duijn CM, Sims R, van der Flier WM, Ruiz A, Ramirez A, Lambert JC. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022 Apr;54(4):412-436. Epub 2022 Apr 4 PubMed.
- Kämäläinen A, Viswanathan J, Natunen T, Helisalmi S, Kauppinen T, Pikkarainen M, Pursiheimo JP, Alafuzoff I, Kivipelto M, Haapasalo A, Soininen H, Herukka SK, Hiltunen M. GRN Variant rs5848 Reduces Plasma and Brain Levels of Granulin in Alzheimer's Disease Patients. J Alzheimers Dis. 2012 Aug 13; PubMed.
- Götzl JK, Brendel M, Werner G, Parhizkar S, Sebastian Monasor L, Kleinberger G, Colombo AV, Deussing M, Wagner M, Winkelmann J, Diehl-Schmid J, Levin J, Fellerer K, Reifschneider A, Bultmann S, Bartenstein P, Rominger A, Tahirovic S, Smith ST, Madore C, Butovsky O, Capell A, Haass C. Opposite microglial activation stages upon loss of PGRN or TREM2 result in reduced cerebral glucose metabolism. EMBO Mol Med. 2019 Jun;11(6) PubMed.
- Katsumata Y, Shade LM, Hohman TJ, Schneider JA, Bennett DA, Farfel JM, Alzheimer’s Disease Genetics Consortium, Kukull WA, Fardo DW, Nelson PT. Multiple gene variants linked to Alzheimer's-type clinical dementia via GWAS are also associated with non-Alzheimer's neuropathologic entities. Neurobiol Dis. 2022 Nov;174:105880. Epub 2022 Sep 30 PubMed.
- Korhonen VE, Remes AM, Helisalmi S, Rauramaa T, Sutela A, Vanninen R, Suhonen NM, Haapasalo A, Hiltunen M, Jääskeläinen JE, Soininen H, Koivisto AM, Leinonen V. Prevalence of C9ORF72 Expansion in a Large Series of Patients with Idiopathic Normal-Pressure Hydrocephalus. Dement Geriatr Cogn Disord. 2019;47(1-2):91-103. Epub 2019 Mar 12 PubMed.
- Rostalski H, Korhonen V, Kuulasmaa T, Solje E, Krüger J, Gen F, Kaivola K, Eide PK, Lambert JC, Julkunen V, Tienari PJ, Remes AM, Leinonen V, Hiltunen M, Haapasalo A. A Novel Genetic Marker for the C9orf72 Repeat Expansion in the Finnish Population. J Alzheimers Dis. 2021;83(3):1325-1332. PubMed.
Other Citations
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
Columbia University
The UEF NEURO-RC, led by Mikko Hiltunen, is a truly remarkable program. We in the field have historically suffered from "biopsy envy." Unlike other diseases that have the advantage of performing detailed cellular and molecular investigations on biopsied samples, we had to rely on postmortem autopsied samples. This program relies on NPH patients who need to have shunts placed for their medical management to take biopsy samples of the frontal cortex. Since these patients often end up having Alzheimer’s disease, these samples can be used to better understand the cellular and molecular underpinnings of the disease.
Make a Comment
To make a comment you must login or register.