Caught amid COVID-19, the organizers of this meeting decided on March 10, 2020, to switch to a virtual format. It offered prerecorded lectures and e-posters, as well as livestreamed discussions, “Meet-the-Professor” audio chats, and a virtual exhibit hall. New data included widely anticipated results of the DIAN-TU clinical trial of solanezumab and gantenerumab, and of robust phospho-tau plasma tests. The online stream also brought new data on a range of trials against tau, α-synuclein, and other targets in age-related neurodegenerative disease, and on pathogenesis and other topics.
New Assay, New Cohorts—Plasma p-Tau181 Looks Even Better
The conference may be virtual, but the data is real. At the online AAT-ADPD 2020, researchers presented their talks and posters in much that same way as they would have in person—albeit without the open question-and-answer time. Up today was a presentation on plasma markers by Kaj Blennow, University of Gothenburg, Sweden. He showed that a new blood immunoassay his group developed for p-tau181 may be the most sensitive yet. It robustly detected a steady rise in plasma levels of the marker as Alzheimer’s disease progressed. Plasma p-tau181 distinguished people with AD from healthy controls and from those with frontotemporal dementia and other primary tauopathies with a high degree of accuracy, Blennow reported. Coming from 1,131 samples, the data corroborate findings recently reported in two Nature Medicine papers. Those studies used a proprietary p-tau181 assay developed by Jeffery Dage at Eli Lilly and Company, Indianapolis.
“It is always comforting when we have slightly different assays that give the same result,” Henrik Zetterberg, UGot, told Alzforum. A co-author on all three papers, Zetterberg said both assays work beautifully. “I didn’t think measuring phospho-tau in the blood would be possible, but this has been super straightforward,” he said.
What has become known as the Dage assay runs on the Meso Scale Discovery platform. Owned by Lilly, it works like an ELISA, using capture and detection antibodies, but electrochemiluminescence rather than colorimetry quantifies binding of the latter for higher sensitivity. This assay is proprietary. Blennow and colleagues developed their own immunoassay on a single molecule array platform. Simoa is generally more sensitive than MSD. They used the same capture antibody as Dage, the AT270 mouse monoclonal, which recognizes phospho-threonine at position 181 of tau. For detection, the Gothenburg test uses the commercially available Tau12. It binds the N-terminal sequence QEFEVMEDHAGT, which begins at amino acid six of human tau. The Dage assay uses an anti-tau monoclonal developed by Lilly for detection. It also binds the N-terminal, but the epitope is just a little bit different, said Zetterberg. Researchers in Japan previously reported a p-tau181 immunoassay, but it reversed the antibody steps, using a total-tau antibody for capture and a p-tau181 antibody for detection. It did not detect p-tau181 in 11 of 15 control samples and four of 20 AD samples (Tatebe et al., 2017).
In Agreement. In the Triad and BioFINDER 2 cohorts, plasma p-tau181 tracks with severity of AD progression. [Courtesy of Kaj Blennow, University of Gothenburg.]
In his AAT-ADPD presentation, Blennow reported that the new assay’s so-called “lower limit of quantification” lies at 0.5 pg/mL. This means that p-tau181 was measurable in all 1,131 samples, even those from healthy controls as young as 23.
By contrast, in their Nature Medicine paper, researchers led by Adam Boxer at the University of California, San Francisco, reported an LLOQ of 1.4 pg/mL for the Dage assay. They were unable to detect p-tau181 in 41 of 404 samples from three cohorts (Thijssen et al., 2020). Similarly, Oskar Hansson’s group at Lund University, Sweden, found that of 589 samples, 20, including two from Aβ-positive individuals, fell below the level of detection using that same assay (Janelidze et al., 2020; Mar 2020 news). Level of detection is a less stringent metric than LLOQ.
Whether this small difference has practical consequences remains to be seen. “The Simoa assay may be more sensitive, but I don’t think it matters all that much, since we have so few samples below the limit of detection,” said Hansson. Zetterberg cautioned that it is hard to compare the analytical sensitivity of the two assays based on their LLOQs, since they are not calibrated or standardized to each other.
What does the new Simoa assay tell us? Thomas Karikari at UGot, working with TharickPascoal from McGill University, Montreal, measured p-tau181 in four different cohorts. For discovery, they tested 19 people with AD and 18 age-matched controls from UGot. For validation they used 226 volunteers from the Canadian TRIAD and 763 from the Swedish BioFinder-2 prospective studies, respectively. The fourth set was 105 people from primary care; they included young healthy controls and people with MCI or AD. The study was a collaboration with Hansson’s group and with Pedro Rosa-Neto’s lab at McGill.
In the discovery cohort, the assay detected p-tau181 in both plasma and serum. “The assay works equally well in both,” Blennow said. Levels were elevated in AD compared with controls, and the same was true in TRIAD and BioFinder-2 (see image above). In TRIAD, people who were Aβ-positive had higher plasma p-tau181 than did young controls, older Aβ-negative volunteers, and people with FTD. Old people, both cognitively normal or with MCI, had more plasma p-tau181 if they were Aβ-positive. People with AD had the highest plasma p-tau181 levels; their mean was 24.9 pg/mL. The marker distinguished AD from FTD, cognitively normal, and MCI with AUCs of 1.0, 0.95, and 0.85 respectively.
BioFinder-2 told the same story. Plasma p-tau181 ticked up across the disease spectrum as diagnosis changed from Aβ-negative normal/MCI, to Aβ-positive normal/MCI, to AD. Plasma p-tau181 distinguished AD from PD/MSA, PSP/CBS, VaD, and bvFTD/PPA with AUCs of 0.82, 0.88, 0.92, and 0.83 respectively.
As was reported in the two Nature Medicine papers, Blennow, too, found that plasma p-tau181 as measured by the new assay was strongly correlated with CSF p-tau181 and with neurofibrillary tangle burden as judged by tau PET; AUCs for these correlations were high, at 0.91, 0.92, and 0.94 for SUVRs in Braak stages I-II, III-IV, and V-VI, respectively. In keeping with the idea that p-tau181 is an early response to Aβ toxicity, plasma levels among tau PET-negative volunteers were higher if the person was Aβ-positive. “This suggests plasma p-tau181 is an indicator of very early brain amyloidosis,” said Blennow.
In a separate talk, Karikari reported data from the 105 people in the primary care setting. P-tau181 was higher in the cognitively normal elderly than in young controls, and higher again in those with mild cognitive impairment. Values were highest in AD samples, where levels discriminated against the young, people over 60, and MCI with AUCs of 1.0, 0.84, and 0.55, respectively. The overlap between the AD and MCI patients suggests that the latter should be selected for specialist care, said Karikari.
To bolster this primary care data, Karikari analyzed 312 samples from the European AddNeuroMed prospective study. This public-private partnership coordinated by Simon Lovestone at King’s College London correlates blood marker data, cognition, and MRI analysis. Here, too, p-tau181 levels were lowest in samples from cognitively unimpaired, higher in MCI, and highest in AD samples. It correlated with clinically diagnosed AD with an AUC of 0.922, which was higher than the 0.830 recorded for neurofilament light, another promising marker for AD and other neurodegenerative diseases. The Boxer and Hansson groups also found that p-tau181 outperformed NfL in distinguishing AD from controls and from people with other neurodegenerative diseases. In fact, layering NfL on top of p-tau181 added no benefit, Hansson reported.
Blennow said this work is slated for publication in the April 22 Lancet Neurology—slightly delayed thanks to COVID-19.
All told, these three latest papers detailing separate analyses of more than 2,000 blood samples paint plasma p-tau181 as a noninvasive, robust, and specific marker of Aβ toxicity. “Plasma P-tau181 may be used as a first line of testing to identify patients likely to be tau-positive when tested by PET or CSF biomarkers, either to distinguish AD from other non-AD neurodegenerative diseases in cases with mild to moderate dementia or to predict future development of AD in cases with mild cognitive impairment,” wrote Hansson and colleagues.
Zetterberg thinks blood p-tau181 is ideal for monitoring responses to experimental drugs. “It almost seems that a successful therapy would have to attenuate p-tau181 levels,” he said. This could be tested in ongoing and even in completed trials. Companies have thousands of plasma samples from prior trials. “You would think they would assay for p-tau181 immediately,” Zetterberg said. He has asked for samples but has not yet received any.
Is a clinical-grade p-tau181 test far off? Zetterberg is convinced that this data is real and will hold up. He said the next step will be a fully automated, sensitive assay that could be rolled out in the clinic for diagnostic use.
Hansson agreed. “We need a reliable assay that works anywhere in the world, akin to the Elecsys or Fujirebio platforms used to measure CSF analytes,” he said. Roche, which sells the Elecsys system, is putting a lot of effort into this new marker, said Hansson. In parallel, the field will need to create a reference material that can be used to standardize tests, perhaps a tau fragment quantified by mass spectroscopy. “We need this type of development now,” said Zetterberg.
Could this push aside Aβ tests? Plasma Aβ42/40 falls by up to 15 percent in people with brain amyloid, a small change that is more difficult to measure accurately than p-tau181, which can more than quadruple. Different immunoassays for plasma Aβ42/40 don’t always agree either (Aug 2019 news). On top of this, immunotherapies that target free Aβ will bind it in blood, skewing plasma analysis. Despite these disadvantages, researchers still think plasma Aβ tests will be useful. “When we develop novel therapeutics, it makes sense to see how they affect amyloid,” said Zetterberg. He and Hansson both emphasized that Aβ changes will precede p-tau181 changes. “When we test very early, plasma Aβ42/40 may still offer an advantage,” said Hansson.—Tom Fagan
A flood of recent data seems to leave little doubt that phospho-tau217 is the better of the soluble tau markers for studying Alzheimer’s disease thus far. At this year’s virtual AAT-AD/PD meeting, Oskar Hansson, Lund University, Sweden, reported that p-tau217 in the CSF correlates more strongly with neurofibrillary tangles and amyloid plaques than does p-tau181 or total tau. CSF p-tau217 climbs higher, and better distinguishes AD from controls and non-AD forms of dementia, as well, said Hansson.
Both 217 and 181 p-tau ticked up in the CSF before tau PET scans became positive, indicating that phosphorylation at these two sites represents a very early response to Aβ. The findings appear in the April 3 Nature Communications and the April 15 Science Advances, the latter paper being delayed due to the coronavirus pandemic.
Separately, in the March 17 Alzheimer’s Research & Therapy, researchers led by Nicolas Barthelémy at Washington University, St. Louis, Audrey Gabelle at Gui de Chauliac Hospital, Montpellier, France, and SylvainLehmann, University of Montpellier, reported that of the two phospho-taus, p-tau217 better correlates with amyloid plaques. Almost without exception, people with AD have more of the marker in the brain than anyone else. Hansson thinks p-tau217 will soon take over from p-tau181 as the go-to tau marker for AD. “While the difference [between them] is not huge, it is consistent, so why use a test that is less accurate?” he asked.
Eric McDade, WashU, agreed. “As the authors suggest, with validation in larger cohorts, p-tau217 may become the gold standard as an AD diagnostic test, particularly if this phosphorylation site can be reliably detected in the blood.” (See full comment below.)
These reports come shortly after a paper last month by Barthelémy and colleagues. It detailed how p-tau217 is the first tau species to rise in the CSF of people with autosomal-dominant AD, edging up some 21 years before the expected age of symptom onset (Mar 2020 news). The data in the new batch of papers pertain to sporadic AD.
A Better Test. Across the AD spectrum, fold changes in CSF p-tau217 (left) are larger than for p-tau181 (right). [Courtesy of Shorena Janelidze, Nature Communications.]
In Hansson’s group, Shorena Janelidze compared levels of p-tau217 and p-tau181 in CSF samples from the Swedish BioFinder study. Both tau species were measured on the Meso Scale Discovery platform, using immunoassays developed by Jeffrey Dage at Eli Lilly, Indianapolis. These two-step ELISAs used antibodies specific to p-tau217 and p-tau181 for capture, but the same N-terminal tau antibody for detection; this should reduce between-assay variability, the authors noted. Janelidze measured both isoforms in CSF from 194 volunteers, of whom 184 had had a flortaucipir PET scan for neurofibrillary tangles and 139 a flutemetamol scan for amyloid. The cohort included 65 cognitively normal controls, 29 with Aβ-positive MCI, 43 with AD, and 57 who had non-Alzheimer’s disease dementia.
Levels of both p-tau forms were highly correlated to each other. That said, p-tau217 showed a higher dynamic range across the AD disease spectrum. It rose eightfold in MCI/AD over Aβ-negative normal controls, where p-tau181 was fourfold higher.
Both p-taus correlated with plaques and tangles as per PET. Again, this was stronger for p-tau217, which covered more “area under the curves” for predicting flortaucipir binding at all Braak stages of tau progression. Likewise, the AUCs for predicting abnormal CSF Aβ42 and amyloid PET were 0.910 and 0.827, respectively, for p-tau217, versus 0.890 and 0.799 for p-tau181. Measuring the ratio to total tau improved these AUCs for p-tau217 to 0.914 and 0.871 and for p-tau181 to 0.914 and 0.859. All told, the data indicate that p-tau217 is a more robust marker of underlying AD pathology.
Janelidze found a similar pattern in a second BioFinder cohort of 330 people comprising normal controls and people with MCI. To corroborate further, the researchers tested CSF from 32 people in Lilly’s EXPEDITION3 trial of solanezumab. Here, too, CSF p-tau217 correlated with flortaucipir binding even more strongly than did p-tau181.
The pattern in BioFinder also held when p-tau217 and total tau were measured with a different pair of immunoassays developed by Janssen Research and Development, La Jolla, California. Again, p-tau217 better correlated with tau PET and better differentiated AD from other neurodegenerative disorders. “Taken together, these findings suggest that increases in CSF p-tau might reflect AD-related tau hyperphosphorylation rather than only increased production and secretion of tau from cells, and that in AD, changes in soluble tau metabolism affect to a larger extent phosphorylation at Thr217,” write the authors.
Unambiguous? In the Montpellier cohort, mass spectrometry shows that p-tau217 (bottom) is consistently higher in the CSF of people with AD than in the CSF of those with other dementias, some of whom (numbers 1 to 3) had abnormally high p-tau181 (top). [Courtesy of Barthelemy et al., Alzheimer’s Research & Therapy.]
Barthelémy and colleagues reported similar findings among a cohort of 50 clinically well-characterized dementia patients in Montpellier. Mass spec analysis of their CSF indicated a higher dynamic range for p-tau217, much like Hansson and colleagues found. This isoform rose up to sixfold in the AD samples compared with non-AD, whereas the 181 form only rose 1.3-fold. The 10 people with AD had consistently more p-tau217 in their CSF than did 40 people with non-AD dementias. Here again, the 217 isoform performed best, since three of the non-AD samples had p-tau181 levels in the AD range, a recipe for misdiagnosis (see image above).
These researchers also compared CSF phospho-tau isoforms in a cohort of 84 people from the Alzheimer’s Disease Research Center at Wash U. This included 51 Aβ-negative controls and 33 Aβ-positive individuals with mild cognitive impairment or AD. While there was considerable overlap between levels of p-taus in the two groups, p-tau217 was higher in almost all Aβ-positive samples, distinguishing Aβ-positives from -negatives with an AUC of 0.961. For p-tau181 the AUC was 0.785.
P-tau217 also tightly correlated with amyloid PET. “The data obtained show that CSF pT217 is a much more highly specific marker than pT181 for detecting both preclinical and advanced AD,” write the authors.
The data from both groups support the idea that phosphorylation of tau at specific sites is an early and specific response to Aβ toxicity, and that it precedes the formation of neurofibrillary tangles. In his ADPD presentation, Hansson drilled deeper into when in the disease tau gets phosphorylated. Niklas Mattsson and colleagues at Lund compared soluble tau and tangle levels in BioFinder. Of 131 people studied, 113 had brain amyloid based on their Aβ42/40 ratio in the CSF. They included 40 people who were cognitively normal, 38 with MCI, and 35 with AD. The remaining 18 were amyloid-negative and cognitively normal.
Mattsson found that of the 18 controls, 17 tested negative for soluble CSF tau and tau PET; one person had both high p-tau217 and p-tau181. However, among the Aβ-positive cognitively normal volunteers, there were already signs of tau pathology. Of these 40, nine, 22, and 28 had elevated total tau, p-tau181, and p-tau217, respectively. Still, only one had tangles in the inferior temporal cortex (ITC) as per flortaucipir binding, suggesting that tangles come later.
Analysis of the MCI and AD groups supported this time sequence. Nearly all had elevated p-tau181 and p-tau217, and 73 percent also had elevated total tau. Three-quarters of those with MCI had tangles in the ITC, while everyone with AD did. Tangles had spread to Braak stage V-VI regions in 32 percent of the MCI subjects and 66 percent of the AD patients.
The data indicate that soluble p-tau emerges before tangles. Looking at this a different way, Hansson compared ITC tangle and soluble tau positivity. He used cutoffs of 1.31 SUVR for the former and 152.6 pg/mL and 119.5 pg/mL for p-tau181 and p-tau217, respectively. For half the cohort the markers were concordant, meaning people tested positive for both. When the markers were discordant, it was almost always the phospho-tau that was abnormal. For example, 28 percent of people tested positive for only p-tau217, but only 1 percent tested positive for only tangles. “The data suggest that people move from normal CSF and normal PET, to abnormal CSF and normal PET, and then to abnormal CSF and abnormal PET,” said Hansson.
How does this tie in with Aβ? Hansson showed that p-tau181/217 turn positive before amyloid PET does, while tau PET turns positive after. Again, this puts phosphorylation before tangles in the pathological cascade. In fact, correlating tau PET with amyloid PET, Mattsson and colleagues found that as amyloid rose, tangles only accumulated if people had already tested positive for p-tau181 (they did not look at p-tau217 in this scenario). “This suggests you need p-tau181 to get tangles,” said Hansson. Mattsson agrees. “There is a very strong relationship between amyloid and tau PET, and it is to a very large degree mediated by changes in soluble tau,” he said. Mediation analysis indicated that p-tau217 and p-tau181 explained 82 and 68 percent, respectively, of the Aβ-driven increase in tangles.
Mattsson stressed that this is all still a bit speculative. “To really prove this chain of events, we’d want to block increases in soluble tau and see if that prevents the transition to tau aggregates,” he said. There are hints from human studies. For example, Mattsson found that people with MAPT mutations, who have no Aβ pathology, also have no p-tau181/217 in their CSF even though they do accumulate a different type of tangle than seen in AD.
This suggests Aβ kicks of a specific tau cascade that begins with these phospho forms. Just recently, data from the DIAN-TU trial of gantenerumab showed that this anti-Aβ immunotherapy dramatically reduced levels of soluble total tau and p-tau181 (April 2020 conference news). “This is a very nice way, with an intervention study, to address the cascade we are suggesting,” said Hansson. “The next step would be to see if the treatment reduces the accumulation of tangles.”—Tom Fagan
In DIAN-TU, Gantenerumab Brings Down Tau. By a Lot. Open Extension Planned
Against classical music and visual backdrops evoking Vienna, where the second AAT-AD/PD conference was to be held, the meeting instead unfolded Netflix-style. In this surreal age of COVID-19, the conference organizers dropped five days’ worth of prerecorded scientific programming all at once, tempting registrants around the world to binge-watch Alzheimer’s and Parkinson’s research symposia like so many episodes of “Stranger Things.” What stood out in this stream of slides, posters, and glimpses of speakers’ home offices? Some important news, actually. Consider this story of how gantenerumab gained a second wind in the Dominantly Inherited Alzheimer’s Network trials unit (DIAN-TU). While the primary outcome posted a null result, gantenerumab turned out to slash not only Aβ, but also tau. Could a higher dose work, after all?
Topline data presented by Randall Bateman, Washington University, St. Louis, suggest that the first two drug arms of the DIAN-TU trial platform—of Lilly’s solanezumab and Roche’s gantenerumab—were not a complete bust. Instead, the analyses finished to date point to nuanced effects of dose, time, disease stage, and biology. To be sure, the data shown at AAT-AD/PD did substantiate the earlier announcement that both therapeutic antibodies had fallen short on the trial’s primary endpoint, the DIAN-TU multivariate cognitive endpoint (Feb 2020 news). What happened? In short, symptomatic participants had descended into moderate dementia even before they could be titrated up to a high dose, whereas asymptomatic participants stayed stable throughout the trial regardless of whether they were on drug or placebo. This left the trial’s main question unanswered.
For solanezumab, a monoclonal antibody targeting soluble Aβ, this indeed marks the end of its exploration within DIAN, the global research network for families with autosomal-dominant Alzheimer’s disease. But all is not lost for gantenerumab, a monoclonal targeting aggregated forms of Aβ. Besides removing amyloid plaques from the brain and normalizing CSF Aβ42, this antibody reversed toward normal the elevated levels of CSF total tau and p-tau181, an AD-specific, pathological form of this neuronal protein. Gantenerumab further stemmed the rise of the general neurodegeneration marker CSF neurofilament light.
“I am very encouraged by the gantenerumab data. The tau and NfL response are good evidence that Aβ is driving these downstream changes,” Colin Masters of the University of Melbourne wrote to Alzforum.
“We are faced with the conundrum of a powerful drug that reaches target and has significant downstream effects, but no clinical effect, in an otherwise well-designed and well-executed trial,” Philip Scheltens of Vrije University Amsterdam summed up the data.
The effect sizes of this biomarker response were so large that they prompted the DIAN investigators and Roche to invite DIAN participants—who have devoted four to seven years of their lives to this trial, depending on when they enrolled—to join an open-label extension. It will explore high-dose gantenerumab therapy for several additional years.
Its goal? To see if sustained gantenerumab therapy near the highest tolerated dose removes both plaques and tangles all the way down to a hypothesized, yet-to-be-defined threshold at which cognition and function might start to benefit. The researchers also want to learn if a longer time on such a high dose gives the brain time to adjust to life without plaques and tangles—that is, to heal. Can a brain newly freed of this pathology cool its inflammation and perhaps restore synapses and circuits?
Staging DIAD. Twelve years—and counting—of observational research participation by families with pathogenic APP and presenilin mutations is continually refining this staging diagram of proteinopathy and downstream changes in this rare form of Alzheimer’s disease. [Courtesy of DIAN-TU.]
The design for this closely watched DIAN-TU-001 intervention trial was informed in large part by data gathered during the DIAN observational (Dian-obs) cohort study started by John Morris and colleagues at WashU, and now led by Bateman. In 2008, DIAN-obs began to characterize Alzheimer’s disease pathogenesis from early adulthood onward in carriers of rare pathogenic mutations in the APP and presenilin 1 and 2 genes, and compare it with their non-carrying relatives (Nov 2008 news). Clinical, cognitive, fluid biomarker, and brain-imaging measurements gathered since then helped build a model of disease progression. It aims to span a 40-year arc from early elevated CSF Aβ42 levels 30 years before a carrier develops symptoms, all the way to full-fledged dementia 10 years after onset (Bateman et al., 2012; McDade et al., 2018).
DIAN scientists used this observational data for two main purposes. They built a mathematical disease-progression model to serve as a quantitative framework for treatment trials in this form of AD. They also tap this observational data to supplement the placebo dataset of DIAN-TU treatment trials so that fewer mutation carriers have to be on placebo during a multiyear trial. (The odds of being on placebo deters people from committing to long clinical trials.)
DIAN-TU Aims. Since 2012, the Dominantly Inherited Alzheimer’s Disease Trials Unit started dosing in three trials and completed two of them. A cognitive run-in phase is ongoing for two trials targeting tau and a primary prevention trial, whose study drugs are yet to be announced. [Courtesy of DIAN-TU.]
Preparations for an ongoing platform of successive DIAN-TU drug trials started nine years ago (Dec 2011 conference news). It engaged families the world over, together with pharma and other companies, academics and their site staff, funders, and other stakeholders, in an intensive public-private partnership. This PPP has thus far planned a series of seven drug arms, of which the solanezumab and gantenerumab treatment arms are the first two. The third arm, of the BACE inhibitor atabecestat, stopped in 2018 due to side effects, and arms four to seven will test drugs targeting tau and attempt primary prevention, respectively.
The solanezumab and gantenerumab trials began in December 2012 as a Phase 2 biomarker study and were later switched to a Phase 3 cognitive endpoint study. They enrolled across disease stages. This means they combined in one treatment group asymptomatic mutation carriers who could be as far as 15 years away from their estimated symptom onset and symptomatic people whose clinical dementia rating was 1 or lower at baseline. The first participant received the first of monthly infusions of antibody or placebo in March 2013; the last one got the final dose in November 2019. On average, the trial lasted 5.2 years. Because of the trial’s “common close” design, where everyone stays in until the last enrolled person gets his or her last dose, the earliest-enrolled completers were in this trial for a whopping 2,372 days, or 6.5 years.
Once Again: Too Little, Too Late?
Importantly, midway through this time period, the Alzheimer’s field at large realized that anti-Aβ antibodies need to be given at much higher doses than initially thought if they are to be effective, and that their ARIA side effect was more manageable than initially thought. As trialists did for the Aβ antibodies aducanumab and crenezumab, and for the A4 trial of solanezumab, DIAN-TU decided to ramp up. Starting in August 2016, they escalated the gantenerumab dose fivefold and, starting in September 2017, escalated solanezumab fourfold. This, however, came late in the trial, and symptomatic participants had already declined significantly.
Seven Years. Over its long course, the first trial in dominantly inherited AD switched from a biomarker to a cognitive outcome trial, and escalated doses of both study drugs. [Courtesy of DIAN-TU.]
The execution of the DIAN-TU-001 trial worked, despite widespread doubt early on whether a long, global intervention study could be done in this rare disease at all. Of 236 participants screened—from both within and outside of the pre-existing DIAN observation cohort—193 were randomized to double-blind treatment. Included in this number were 49 noncarriers randomized to placebo; they served as “cover” so no one would have to find out their mutation status. This generated treatment groups of 52 mutation carriers each for gantenerumab and solanezumab, and a pooled placebo group of 40 carriers.
Of those groups, 39, 36, and 30, respectively, completed the trial. This comes out to 7 percent dropout per year. Half of those left because their dementia had advanced beyond the point where they could deal with the infusions and assessments. “The overall dropout rate was amazingly low,” wrote Scheltens. Much shorter Alzheimer’s trials typically lose a third of their patients before the end. Remarkably, during the arduous regimen of monthly infusions plus periodic brain scans, cognitive tests, and fluid donation required, the participants’ compliance rate was 99 percent.
Alas, success on the trial’s main question was not to come. The randomization worked—i.e., the groups started out balanced across age, ApoE genotype, cognitive and clinical baseline, and other factors measured at baseline—but neither gantenerumab nor solanezumab met the primary, i.e., the DIAN-TU multivariate, endpoint. The difficulty was compounded by the realization, during analysis, that the trial data did not meet some of the assumptions previously made in the disease-progression model. Put simply, the data did not fit the model and, as a result, permitted no conclusions to be drawn about the effect of the high dose.
On the primary endpoint, neither treatment group did significantly better than placebo by year four, the last time point at which cognitive and biomarkers were both assessed. (At year five, cognitive but not biomarker data were assessed, and the groups were smaller due to the dropouts.) Overall, both placebo and drug arms declined.
Plotting each of the endpoint’s four component tests individually revealed that one test—logical memory—went against the three others. Participants improved on it year-on-year, showing an unexpected practice effect that was not apparent in the DIAN observational cohort data. The trial participants might have been learning because they took this test every six months, more frequently than it had been administered during the DIAN observation study, Bateman said. On two clinical measures shown, the CDR-SB and functional assessment scale (FAS), combined groups declined in nearly overlapping curves. DIAN-TU scientists are still analyzing additional outcome measures.
An aha! moment came when the scientists broke out the people who were presymptomatic at baseline from those who were symptomatic. The former improved greatly on the logical memory test, whereas the latter did not. On a second test, called Digit Symbol Substitution, presymptomatic participants also improved, but symptomatic people declined. This test was also performed more frequently in the trial than in DIAN-obs. And on the MMSE, which was given annually, presymptomatic people stayed largely stable, near 30, whereas the symptomatic ones slid from 26 to around 20. Overall, separating symptomatic and presymptomatic participants revealed, at a glance, a stark difference between them on all tests used. Symptomatic participants started off much worse and declined; asymptomatic participants started nearer the tests’ ceiling or normal range, and stayed there.
“I think mixing the asymptomatic with the symptomatic effectively cut the power of the study by half. The numbers of cases were too small to allow for a workable cognitive readout,” Masters wrote to Alzforum. On the CDR-SB and FAS, too, all symptomatic groups declined and all presymptomatic groups stayed stable.
To Scheltens’ mind, the absence of evidence on the cognitive endpoint in this trial is not evidence of absence. He thinks that treated participants in this trial might have had slight shifts in cognition, or indeed function, that went undetected. Scheltens suspects the tests used were too “old school” to pick up subtle changes in this relatively young population even if they were sensitive to decline in the observational cohort. He hopes the DIAN-TU-001 data can be mined to look for those shifts. “I always think of the adage, ‘every trial is as good as its outcome measure,’” Scheltens wrote to Alzforum (full comment below).
Part of the problem with the trial data arises from the disease-progression model. It constitutes the statistical construct for analysis of the primary endpoint data in both trial arms. The model was fitted to the DIAN-obs data. To compensate for limitations inherent in running treatment trials in this rare disease, the model made assumptions, for example that performance on all tests would decline, or that variance would be constant across presymptomatic and symptomatic participants, and also about when symptoms would likely start.
“However, and unfortunately, our trial data did not match all assumptions of the model,” Bateman said. The placebo group improved on some tests; measures did not change longitudinally the way cross-sectional analysis had suggested, and asymptomatic and symptomatic carriers had different degrees of variance on the MMSE.
On safety, the news was good. Side effects in the DIAN-TU-001 trial were as in prior trials with these antibodies. Nothing new cropped up. Solanezumab did not cause ARIA. Gantenerumab brought with it more ARIA-E and ARIA-H seen on MRI. Most instances of ARIA resolved spontaneously and were asymptomatic, or they were mildly symptomatic such that the symptoms came up only once participants were specifically asked. “We were very pleased with the great tolerability of gantenerumab in these subjects. It raises the question of whether we could have gone higher with the dose,” Rachelle Doody of Roche said in an accompanying discussion.
Biomarkers to the Rescue?
The U-turn toward hope came with the biomarker analysis. The near-total completion rate of assessments in this trial generated a plethora of fluid and imaging data. At AAT-AD/PD, Bateman first presented prespecified analyses of each drug arm compared with placebo. As was expected, gantenerumab showed target engagement via a statistically significant reduction on PiB PET. At 0.64 SUVR, this reduction was large, and similar to the reduction previously reported for high-dose gantenerumab in a LOAD open-label extension (Dec 2019 conference news).
On downstream disease-modification markers, FDG PET and thickness of the precuneus, an early affected part of the cortex, were not significantly different in the combined asymptomatic/symptomatic groups with either antibody. Bateman did not report tau PET data at AAT-AD/PD because DIAN-TU added this marker midway through the trial, hence has few scans taken at baseline for comparison.
In CSF, the Aβ42/40 ratio was up significantly, and CSF total tau and CSF pTau181 were down by nearly a third. These three changes all indicate a reversal toward normal levels, and came in at p values of below 0.001. CSF neurofilament light chain (NfL) showed less increase on gantenerumab than placebo, at p=0.024.
How about solanezumab? It also engaged its primary target, as evidenced by a steep increase of total CSF Aβ42; however, of the downstream markers, only CSF NfL was significantly different between drug and placebo, and its change pointed toward worsening on drug, with p=0.017. CSF total tau and pTau181 did not budge.
To show how gantenerumab engaged its target over time, Bateman showed PiB PET scans taken at baseline and at one, two, three, and four years. For presymptomatic and symptomatic carriers grouped together, amyloid burden diverged at one and two years and, once participants went on the high dose, the gap widened. By year four, the effect size of amyloid reduction, expressed as percent relative risk, was large—209 percent reversal toward normal.
As was the case for the cognitive data, breaking open the PiB data by presymptomatic and symptomatic groups revealed just how different they were at the start of the trial. Unlike for the cognitive readouts, however, both presymptomatic and symptomatic participants responded to gantenerumab with amyloid reduction. Expressed in centiloids, presymptomatic carriers started out near 30; those on placebo added some 10 centiloids, whereas those gantenerumab lost 10, for a difference of 20 centiloids. Symptomatic carriers started out around 85 centiloids; placebo recipients went up above 100 and gantenerumab recipients down to about 65. Mutation noncarriers posted zero centiloids throughout the trial.
“When you look at the PET Aβ loads in these two groups, you see a large baseline difference, which affects cognition more than biomarkers in terms of response to intervention. The lowering effect on Aβ load is very convincing, but not enough to expect a cognitive benefit. I would have expected to see a cognitive benefit if they had got it down to 30 to 40 centiloids,” Masters wrote to Alzforum.
The Aβ42/40 ratio, which reflects active deposition, returned toward normal on gantenerumab and worsened on placebo, Bateman reported.
The bigger surprise was the strength of the data on downstream markers. At AAT-AD/PD, Bateman showed that both tau measures reversed direction toward normal in people on gantenerumab, posting at least 30 percent difference between drug and placebo by year four. In people on placebo, both total tau and p-tau181 rose year-on-year from baseline; on drug, both decreased year-on-year. In noncarriers, total and p-181 tau stayed unchanged. Commentators agreed that these changes are large and likely biologically significant. Neurofilament light chain, considered a generic indicator of active neurodegeneration, rose more in carriers on placebo than on gantenerumab but stayed largely stable in noncarriers, for a group difference of 10.8 percent. Additional downstream markers remain to be analyzed for differences by disease stage.
Because gantenerumab engaged its target and moved several downstream markers of Alzheimer’s disease, DIAN-TU and Roche are inviting all members of this trial—regardless of whether they had been on gantenerumab, solanezumab, or placebo—into an open-label extension study. The OLE is currently approved for three years. Bateman hopes that if continued biological improvements are seen, with trends toward clinical and cognitive benefit, it will run longer.
This OLE will be exploratory, i.e., have no placebo group. But it will offer the participants access to high-dose gantenerumab. Bateman told Alzforum that mutation carriers will be titrated up to a goal of 1,020 mg per infusion, the dose in Roche’s ongoing Phase 3 GRADUATE program for mild late-onset AD. Tolerability and ARIA allowing, the dose might go even higher. “Due to the late increase in dose, we did not completely normalize amyloid load in the groups,” Bateman said. “I believe in the OLE, they will get there,” Masters said.
What’s to be gained? Some answers, and perhaps some clinical benefit, after all. Will high-dose gantenerumab reduce amyloid plaques in these DIAD families below the threshold of positivity? If so, will downstream markers of AD return to normal, as well? And will that help? In other words, will some symptomatic carriers stabilize or get better? And will the presymptomatic people continue to stay well?
DIAN-TU researchers can compare results from this OLE to disease progression as observed in the DIAN-obs cohort. Doody emphasized that because this OLE is not a new trial, it will not yield definitive answers. But even so, it can contribute important information to the package of converging evidence from multiple sources that will ultimately decide gantenerumab’s future as an Alzheimer’s treatment.
Besides tracking cognition, function, and the biomarkers that were part of the blinded DIAN-TU trial, the OLE can add markers to assess possible effects downstream of amyloid reduction. This could include, for example, CSF sTREM2, YKL-40 and PET markers of glial activation, neurogranin and diffusion tensor imaging or other markers of neurodegeneration, and SV2A, NPTX2, or other markers of synaptic health.
After years of infusions, and travel to clinics for scans, tests, and needle pricks—all for a seemingly null result—do the DIAN participants want to continue? Some among them have given five years of their lives to a placebo, and this will be their first chance to get a drug that moves amyloid and tau. For the open-label extension, all participants need to find out their mutation status, as it would be unethical to expose noncarriers to an investigative medicine. In private, DIAN participants debate this question with some anguish.
And yet, they are stepping up again. In force. “The U.K. participants are very interested. They see this OLE as a logical progression and are willing to find out their mutation status to go on it,” Catherine Mummery of University College London wrote to Alzforum. “Our DIAN-TU participants were very disappointed when the trial was pronounced ‘negative,’ but are hopeful about this recent news and happy for the possibility of participating in the OLE,” wrote Bill Klunk from the University of Pittsburgh School of Medicine. The same feedback came from other sites in in the United States and other countries. Said Bill Brooks of Neuroscience Research Australia, who leads the DIAN-TU site in Sydney, “We plan to begin the OLE as soon as the COVID-19 situation allows.”—Gabrielle Strobel
Confused About the DIAN-TU Trial Data? Experts Discuss
At the virtual AAT-AD/PD Focus meeting held April 1 to 5, clinicians and funders involved in the Dominantly Inherited Alzheimer’s Network Trials Unit (DIAN-TU) fired up their home computers to discuss results from the first DIAN-TU treatment and prevention trial of Eli Lilly’s monoclonal antibody solanezumab, and Roche’s gantenerumab. DIAN-TU’s principal investigator, Randall Bateman of Washington University, St. Louis, had presented topline data analyses of the primary outcome, which was head-scratchingly negative. He also presented the first analyses of several of the trial’s biomarker measures, which were robustly positive (for a summary, see Apr 2020 conference news). What does it all mean? Should a trial this small and heterogeneous not have aimed for a Phase 3 cognitive outcome? What’s to be learned? Below are excerpts from this conversation, edited for brevity and clarity.
Nitsch: DIAN-TU increased the dose because the whole field was learning that high-dose exposure is important. Do you believe gantenerumab would have had a cognitive benefit if patients had gone on the high dose in the first two years, before they declined so much?
Doody: That the patients who were asymptomatic to start with did not get on the high dose until halfway through the observation period is a real detriment. Those who were symptomatic at the start did not get onto the high dose until they were already down into the moderate range. So the clinical question is unanswered. We hope that the symptomatic patients would gain clinical benefit if the theories of AD are correct that amyloid and tau are important drivers to the clinical syndrome. We were unable to test a long-enough, high-enough exposure in this study.
We were very pleased at the great tolerability of gantenerumab in these subjects. It raises the question of whether we could have gone higher with the dose.
Nitsch: It was gratifying to see all biomarkers that were presented moving toward normal. Brain amyloid down, CSF Aβ42 up, tau and p-tau down, and the NfL increase prevented. This shows the biology of the antibody is working. The whole field is witnessing that antibodies designed to remove amyloid do their jobs and are followed by other biomarkers going in the right direction. Why did this not translate into clinical benefit? Is there a threshold we need to hit? Do we need to go down to zero? Or is it dose exposure over time?
Bateman: We can’t answer this with the data from the trial. It’s still an outstanding question whether there is a threshold below which we need to get the amyloid before we get a cognitive/clinical benefit, or whether it is simply a time effect. And there’s a third dimension: the stage of disease at which this happens. In our trial, we can’t answer the high-dose question in early symptomatic stages because by the time we achieved the high dose, people were in advanced clinical stages. We have this open question: Could high dose have worked at an earlier stage of dementia? We could not test that.
Your threshold question we can approach in the exploratory open-label extension. What happens if you completely remove amyloid plaques, when people reach normal levels? In the duration we had participants on high dose in this trial, we did not get the majority back to normal yet. But in the OLE, we aim to get there.
On the dimension of time, we obviously need time to determine whether there is that effect. The OLE offers that additional time. It is possible that the biological effect of the antibody and the clinical benefit are separated by a delay, that you need to treat for a certain period of time for the brain to recover or stabilize in a way that manifests as a cognitive or clinical signal. I think some of the data from other trials, and our trial, suggest that you may need a duration to see downstream effects.
Yaari: About time and thresholds, Lilly has a lot of experience with prior solanezumab studies in mild AD populations. The lower dose did not hit primary in the EXPEDITION trials in sporadic AD, but there was a trend favoring solanezumab. In DIAN, unfortunately we averaged only one year at the higher dose. I wish we had more time at the high dose. The solanezumab data here is difficult to interpret, given the very small sample size and the wide variance within the disease-severity spectrum. We have asymptomatic and symptomatic patients all being tested together in the same model, and not all model assumptions were met. We will dive more into the data.
Nitsch: In clinical practice we use thresholds. We try to lower blood pressure to a certain threshold, we try to lower blood glucose to a certain threshold, we try to lower blood lipids to a certain threshold. Is that something we will see for AD as well? Will we have a centiloid value for amyloid PET, or for CSF or blood Aβ42 that is our target for clinical treatment?
Ryan: Those are unanswered questions, but certainly, the one successful trial we had on AD prevention is the SPRINT MIND trial. There, lowering blood pressure to 120 rather than 140 had cognitive benefit (Jan 2019 news). We may see the same thing with amyloid lowering.
Snyder: We do not know exactly what those measures will be. Maybe amyloid, maybe tau, maybe NfL, maybe something that has not yet been measured here. It is intriguing that with a drug that targets Aβ, we see changes in tau, NfL, and likely other measures that we might also be able to look at. Thinking about the future, maybe we will have a set of markers where we can target a threshold and later see a benefit.
Nitsch: Is the open-label extension looking at thresholds or a time frame? At the end of the day, it’s about neuronal integrity and synapse function, and those may take longer than the movement of the markers.
Doody: To answer this, we need converging data sources. We are pleased to extend exploration of this trial into OLE, but it is not a new double-blind trial that has been designed to answer these questions. We will gather information from the continued exposure of these patients, but we won’t get definitive answers. This OLE is exploratory. We hope to learn: Can we drive those biomarkers further in right direction? Can we cross the thresholds everyone is talking about? And will that correlate with a clinical signal? But without a placebo control, this OLE cannot give the definitive answer. We do have a large Phase 3 program on gantenerumab, in which we measure amyloid and tau PET and multiple biomarkers. We will need information from multiple sources to finally understand what it is that we need to achieve, by stage of disease, and how do we personalize this to each individual.
Nitsch: What lessons are we learning from this data as we design future AD and neurodegeneration trials?
Bateman: We have already made some modifications to our NextGen tau trials based on the experience with our three amyloid-based trials of solanezumab, gantenerumab, and atabecestat. We learned that it’s helpful to have parallel approaches to a target, and will continue that with tau by way of three different tau-based drug arms.
We learned that with the number of people enrolled in these trials, we can obtain clear biomarker results, however, the cognitive results are the big question for us. So the NextGen tau trials are starting with a clear outcome on biological engagement on the biomarkers.
We learned that symptomatic and asymptomatic people—though models can adapt and put their data together—behaved distinctly differently in the trial. The asymptomatic people in our trial did not decline. If you were asymptomatic, it was good to be in the trial, regardless of what drug arm you were in. These people were perfectly stable over four years. That’s an important lesson. It affects our design in DIAN-TU, but should be noted by other people who are designing prevention trials. The performance in a trial on cognitive measures may not mirror what we see in observational studies. The kinds of tests we administer in obs studies, the frequency at which they are administered, the results we obtain and the models that we build off those observational studies may not always directly apply to trial-level data, because trials are fundamentally different than observation studies. That is an important lessons learned.
Nitsch: What anti-tau treatments will be evaluated in Next-Gen DIAN trials?
Bateman: We want to bring in three classes. First, monoclonal antibodies that target specific forms of tau to prevent either its toxicity, aggregation, spread; second, gene-based treatments; third, small molecules that prevent or reverse aggregation. We have proposals under review at NIH and are already enrolling into a cognitive run-in to launch those trials in preparation for those drugs when they are ready.
Nitsch: Why don’t the data in the DIAN-TU trials mirror what was seen in the DIAN-observational study?
Bateman: I showed a case example in logical memory. Depending on how the test is given, and how often, there can be a strong learning effect. Just giving it more often can change how it performs. Other tests, though, seem to behave and were administered similarly to the observational study. So you can hypothesize that simply the people coming into the trial, the expectation they have, were by chance alone different. For example, the asymptomatic group had very little decline in the four years they were being monitored. A few did begin to decline in the fifth year, and that was observable, but very little. We hope in the OLE to see what happens when we continue to follow them. We expect that eventually these mutation carriers will decline, and the question is, do they decline at the same rate as carriers in DIAN-obs?
Some measures, both clinical and cognitive, did match well. For those, the DIAN-obs and the DIAN-TU placebo data were comparable, and the level and amount of decline of the two populations were similar. The devil is very much in the details of the specific test, how it is being given, the population that comes in, and how it is being analyzed.
Nitsch: Why did solanezumab seem to increase severity of symptoms?
Yaari: We do not know. We were surprised. But do not draw broad conclusions. It’s a small dataset, with caveats. We have a wealth of data from EXPEDITION.
Nitsch: Are the antibodies targeting the right Aβ species? Solanezumab and gantenerumab give us a comparison of two antibodies that have different binding profiles.
Yaari: At this time, we do not know anything definitive about targeting the different amyloid species. The amyloid hypothesis is alive and well. The prior experience we have in mild LOAD with solanezumab shows a trend toward benefit, so soluble Aβ could still be the right target.
Bateman: The acid test is the trial. If the drug has a target, and that target is engaged in the CNS of patients, and the patients benefit, that shows the target is useful. Making patients better is the practical evidence that validates a target.
Doody: The DIAN-TU trial was not a head-to-head comparison between gantenerumab and solanezumab. It was two trials that shared a placebo group. Different sites did different studies. There are many monoclonal antibodies directed against Aβ, and utility of each of them needs to be tested in its own trial.
Nitsch: What is the feedback from families to these study data?
Snyder: The families increasingly recognize the complexity of their disease. When DIAN-TU investigators recently shared the data with the participating families, there was overwhelming interest in the OLE. The families were clearly looking for the data to make their decision.
Nitsch: What does NIH think about this data with regard to funding future trials?
Ryan: NIA is invested. We’ve gotten a significant increase in funding in the last few years, and now have over 200 ongoing clinical trials. We are committed.
Nitsch: Will all raw data of both trials be made available in an unrestricted manner to researchers in academia and other companies?
Bateman: Data will be made available. The goal of a public-private trial is to maximally inform the field. In DIAN, we have to avoid self-identification and accidental discovery of one’s mutation status. In our observational study we use a procedure where people request data and it goes through an agreement process that has a very high approval rate for qualified investigators. We usually grant data requests within a few weeks. We will model DIAN-TU sharing on that, keeping in mind that trial integrity poses some added issues. We have to keep the trial’s primary aims intact. For example, sharing data before the end of the trial could threaten that. We will share the data in a way that meets the primary aims and protects the participants’ information so they or their relatives are not at risk. That is why we do not just post the data on the internet for everyone to browse. We have a lot of experience with this in DIAN-obs, where we have fulfilled more than 100 data requests from outside of DIAN, and will build on it for DIAN-TU.
Nitsch: As scientists we are learning tremendously from the DIAN-TU study today. In this sense this is not a failed trial. It is an exceptional public-private partnership.
Doody: DIAN is groundbreaking. The way industry, academia, NIH, philanthropy were able to collaborate was an immense advancement.
Yaari: This study, with its high level of data integrity, exemplifies how academia and industry successfully collaborate to address an unmet need.
Ryan: The execution of this trial was excellent, with impressive enrollment and completion over nearly seven years. While the primary endpoint showed no benefit, the data is providing invaluable information that advances our knowledge of the disease, and the biomarker data raise biological questions that need further study.
Bateman: This partnership between people from all over the world has been transformational from a scientific and clinical trials standpoint. The level of engagement, cooperation, helping each other, is extraordinary. That may not come out from the data we are showing today, but I want to share with the community how every team has rallied to the cause and come together to make this trial happen as best it can. That we together got this admittedly very challenging trial completed successfully is a testament to the entire field.
Active Tau Vaccine: Hints of Slowing Neurodegeneration
Because cognition in Alzheimer’s disease declines as tangles spread, and tau research tools are finally in hand, researchers are increasingly homing in on tau immunotherapy. At the second biannual Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held virtually from April 2 to 5, speakers gave updates on two active tau vaccines currently in clinical trials and on passive tau immunotherapies still in preclinical research. For the active vaccines, researchers are evaluating safety and immunogenicity. One of them, AADvac1, reportedly slowed neurodegeneration biomarkers in Phase 2. The other, ACI-35, elicited a weak immune response in people, and needed to be redesigned to boost immunogenicity.
Each tau immunotherapy covered in this story targets a different form of pathologic tau: N-truncated, phosphorylated, oligomeric, or acetylated. Researchers are unsure which of these, if any, is the main culprit. “What is the best tau therapy? Only clinical trials will tell us, and all approaches have to be evaluated,” Luc Buée of the University of Lille, France, wrote to Alzforum.
So far, the passive tau immunotherapies ABBV-8E12 and gosuranemab have failed to budge progression in the primary tauopathy progressive supranuclear palsy, but are still in trials for Alzheimer’s disease (Jul 2019 news; Dec 2019 news). Other anti-tau antibodies, like semorinemab, zagotenemab, and JNJ-63733657, are in early stage trials and did not post results at AAT-AD/PD.
Did Neurodegeneration Slow Down?
Only two active tau vaccines are currently in trials. The furthest along is ADDvac1, developed by the biotech company Axon Neuroscience in Vienna. It is based on work from Michal Novak, then at the Slovak Academy of Sciences in Bratislava. Novak reported that truncated forms of tau could damage synapses and seed neurofibrillary tangles, and founded Axon Neuroscience to develop tau therapies (Feb 2013 news). The ADDvac1 peptide elicits generation of antibodies that recognize truncated monomeric tau, as well as truncated oligomeric or aggregated tau, more strongly than full-length monomeric tau, according to company CEO Michal Fresser. “The antibodies are selective toward pathological tau species,” Fresser told Alzforum.
At AAT-AD/PD, Fresser presented results from the two-year Phase 2 ADAMANT trial. It enrolled 196 people who had mild AD by NIA-AA criteria, along with medial temporal lobe atrophy by MRI or, in a few cases, cerebrospinal fluid Aβ and tau levels consistent with AD. Fresser did not give a breakdown of how many participants had MRI and/or lumbar punctures at baseline. Participants were from eight European countries, with an average age of 71 and MMSE of 23. They received subcutaneous injections of 40 μg AADvac1 or placebo once a month for six months, followed by quarterly booster doses thereafter. Altogether, participants received 11 doses of vaccine and were followed for two years.
Initially, there were 117 participants in the active and 79 in the placebo group. About 17 percent dropped out, leaving 100 active and 63 placebo by the end. Fresser noted that he had planned for 25 percent dropouts, so the study remained well-powered. The primary outcome was safety, which was met. The vaccine was well-tolerated, with no difference in adverse events between the vaccine and placebo groups except for more injection-site reactions in the former. A secondary outcome, immunogenicity, was also positive, Fresser claimed. Throughout the trial, participants maintained about 1.5 μg/ml antibodies, which Fresser called a robust immune response to the vaccine. Most participants produced antibodies with an affinity for tau of 1 nM or better, similar to the affinity of tau monoclonal antibodies. These were the only two parameters Fresser showed to characterize the immune response. However, in an email to Alzforum, Fresser said that, based on analysis of a prior Phase 1 trial, these antibodies are mainly of the IgG1 isotype and bind to aggregated tau in tissue slices from AD, PSP, CBD, and FTD brains. They also inhibit neuronal uptake of pathologic tau in cellular assays, Fresser wrote.
For exploratory biomarkers, Axon researchers report a marked effect on plasma NfL, which is thought to flag neurodegeneration. In normal aging, plasma NfL has been shown to rise about 14 percent over two years, while in mild AD, it goes up by 24 percent (Mattsson et al., 2019).
The placebo group in this trial resembled these observational data. Their plasma NfL rose 28 percent, or 4.9 pg/ml, over the course of the study. In the treatment group, plasma NfL rose 13 percent, or 2.1 pg/ml, similar to the rise seen in aging. The difference between the groups appeared mostly during the second year of treatment, when NfL levels stabilized in the vaccinated participants. The difference between the groups was statistically significant, with a p value of 0.004, and fairly large, with a Cohen’s d of 0.48.
The treatment group posted no overall cognitive benefit over placebo. Because tau imaging data associate tangle burden with cognitive decline more tightly in younger than in older people (Apr 2018 conference news), the researchers ran a preplanned subgroup analysis of participants 67 or younger. In this comparison of 32 vaccinated people versus 11 on placebo, cognitive decline slowed by 42 percent on the CDR-SB, 31 percent on the MMSE, and 26 percent on Activities of Daily Living in the treatment group, Fresser said. The finding was not statistically significant.
This younger subgroup also posted larger biomarker changes than the full cohort. Their reduction in plasma NfL was twice that of the full group, and cortical atrophy on MRI scans slowed by 47 percent, a statistically significant result with a Cohen’s d effect size of 0.91.
The study collected CSF from few participants. Fresser presented CSF data on 20 people on treatment and seven on placebo, who were not stratified by age. Both p-tau181 and p-tau217 dropped in the treatment group, the former by about 5 pg/ml, the latter by 50 pg/ml; these markers stayed stable in the seven on placebo controls. Total tau held steady in the treatment group; it rose in controls by about 60 pg/ml. Fresser did not specify the initial concentrations of CSF p-tau and total tau. The CSF changes were not statistically significant. In another small substudy of 20 people, diffusion tensor MR imaging suggested better white-matter integrity in 13 vaccinated participants compared with seven controls.
“AADvac1 showed a highly significant impact on neurodegeneration, as measured by plasma NfL and supported by an effect on CSF tau, p-tau, and DTI,” Fresser concluded. He believes the data indicate a disease-modifying effect, particularly in younger AD patients. Axon Neuroscience is planning a Phase 3 trial that will run for 24 to 30 months.
Optimizing Immune Response
Researchers at AC Immune in Lausanne, Switzerland, approach tau immunization differently. Marija Vukicevic described the company’s liposomal supra-antigen vaccine approach, which binds a synthetic peptide eight to 60 amino acids long to a liposome. The peptide can be conjugated to the bilayer in such a way as to stabilize a particular shape, producing a conformation-specific immune response. The liposome also holds adjuvants to boost the immune response.
To induce tau antibodies, the researchers used a synthetic peptide based on human p-tau396/404, although Vukicevic did not say what region or conformation (Jun 2012 conference news). The resulting vaccine, ACI-35, preserved motor abilities and extended survival of mice carrying a P301L mutation (Aug 2014 news).
At AAT-AD/PD, Vukicevic reported results from the Phase 1b trial. The vaccine was well-tolerated but elicited a weak immune response, and booster shots had little effect. So the researchers redesigned the vaccine, adding a second adjuvant plus an epitope that activated the HLA-DR receptor on T-cells.
In rhesus monkeys, this second-generation vaccine, ACI-35.030, produced a stronger immune response. Over six months, vaccination with ACI-35.030 generated 50 times as many antibodies as did ACI-35, and booster shots added a more robust effect. As with ACI-35, antibodies generated by the new vaccine were specific for p-tau over tau, showing about 100-fold selectivity in rhesus monkeys, and they recognized paired helical filaments extracted from AD brain.
In collaboration with Janssen, AC Immune is testing this vaccine in a multicenter Phase 1b/2a safety and immunogenicity study in AD patients.
Pinning Down Toxic Tau
One roadblock for tau immunotherapies is that scientists do not yet know which form of tau is most toxic. Different groups are chasing distinct forms, and many approaches are wending their ways through preclinical studies. At AAT-AD/PD, Alice Bittar of the University of Texas Medical Branch presented data on monoclonal antibodies specific to tau oligomers. These so-called TOMAs lower oligomeric tau, but not monomeric p-tau or neurofibrillary tangles, in mouse models of tauopathy. In middle-aged tau mice, TOMA treatment improved cognition (Castillo-Carranza et al., 2014; Castillo-Carranza et al., 2014; Castillo-Carranza et al., 2015).
How about in old mice? Working with Rakez Kayed at UTMB, Bittar characterized two clones, TOMA1 and TOMA3, in 1-year-old JNPL3 mice and 2-year-old hTau mice. She injected 120 micrograms TOMA or control IgG into their tail veins, then tested cognition three or four days later.
Results varied according to the specific antibody clone, Bittar reported. TOMA1 improved JNPL3 mouse performance in the Y-maze and novel-object-recognition tasks, while TOMA3 helped hTau mice. Immunohistochemistry of the mouse brains offered a rationale, as TOMA1 lowered tau oligomers in JNPL3 mice, but not hTau animals, while the opposite was true for TOMA3, Bittar said.
These clones have different staining patterns in postmortem human brain samples. TOMA1 reacts most strongly to Parkinson’s brain, less to AD, and least to PSP. TOMA3, on the other hand, binds to AD brain but not to the others, suggesting it may be specific for the paired helical filaments of 3R/4R tau that accumulate in that disease. The data indicate that tau antibodies are specific to particular tau proteopathies, Bittar said. She proposed that certain diseases might require a combination of therapeutic tau antibodies. Kayed’s group is now testing combinations in mouse models. They are also humanizing two TOMA clones for human trials, but Bittar did not say which ones.
Also at AAT-AD/PD, Buée discussed a different form of pathologic tau. His group analyzed Alzheimer’s brain homogenate by mass spectrometry and found it was enriched for an N-truncated version of tau that starts at Met11. This amino acid is also acetylated. Further research determined that acetylated Met11-tau is only present in AD brain, not other tauopathies. It occurs in insoluble tau fractions along with paired helical filaments, and is found in tau transgenic mouse brain.
Buée injected a viral vector carrying either Met11 or full-length tau into young Thy-Tau 30 mice. Met11-tau potentiated tau pathology more potently than did full-length tau, boosting the number of hippocampal neurons containing tau-positive inclusions by about 50 percent. This confirmed its toxicity. The researchers then generated an antibody to Met11 tau, and injected 10 mg/kg into Thy-Tau22 mice seven times over the course of five months. This treatment squelched accumulation of insoluble tau.
Met11-tau may represent a new target for immunotherapy, Buée suggested. He is looking for a company to partner with to bring the approach to clinical trials.—Madolyn Bowman Rogers
Non-Aβ, Non-Tau Drugs Tweak Markers, Cognition in Alzheimer’s, Huntington’s
As the Alzheimer’s field branches out its therapeutics search beyond Aβ and tau, some of those approaches are showing glimmers of promise. The second biannual Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held virtually April 2 to 5, featured some new data on therapies that target neuroinflammation, synaptic signaling, epigenetic regulation, and the cortisol stress response. The candidate drugs nudged cognition or biomarkers in small, early stage trials of Alzheimer’s or Huntington’s patients, with larger studies planned. Makers of the nutraceutical drink Souvenaid presented three-year data from a recently concluded trial. Cognitive benefits previously reported at two years had grown, suggesting a sustained effect. Some of these approaches could broaden the therapeutic pipeline for neurodegenerative disease.
Nutrient Drink: Benefit Accrues Over Time Souvenaid is a medical food formulated as a once-a-day, yogurt-like drink. It contains Fortasyn Connect, a combination of fish oils, vitamins, and other nutrients believed to support synaptic health. The drink is made by Nutricia of Danone Research. Several small trials suggested a possible cognitive benefit when taken early in the course of AD. To evaluate this lead, the LipiDiDiet trial enrolled 311 people with prodromal Alzheimer’s disease and a mean age of 71. After two years, participants who drank Souvenaid performed no better than controls on the primary outcome measure, the Neuropsychological Test Battery, but had declined less on the CDR-SB and their hippocampi had shrunk a little less (Nov 2017 news). An independent analysis confirmed that Souvenaid curbed cognitive decline in these trial participants by a third (Nov 2018 conference news).
At AAT-AD/PD, LipiDiDiet study coordinator Tobias Hartmann of Saarland University in Homburg, Germany, reported three-year CDR-SB and volumetric data from LipiDiDiet. The third year represented an optional, blinded extension to the initial two-year trial. This far out, 45 participants remained in the treatment and 36 in the placebo group, less than a third of the starting enrollment.
At three years, the cognitive benefit on the CDR-SB widened for those taking Souvenaid, with decline slowing by 45 percent. Likewise, the rate of hippocampal atrophy diverged further between the groups, slowing by 33 percent for those on Souvenaid. The Cohen’s d effect sizes were 0.31 and 0.27, respectively, for CDR-SB and hippocampal atrophy. Both were significant at the 0.01 level or better.
These effect sizes are similar to that seen in the EXPEDITION 1 and 2 studies of solanezumab, where the antibody slowed cognitive decline by a third in the subgroup of participants with mild AD (Jan 2018 news).
“The benefits observed at the two-year intervention were sustained and amplified over three years, with a remarkable magnitude of slope and effect size,” Hartmann said in his talk. He noted that LipiDiDiet is now the longest intervention trial in prodromal AD, with six years of placebo-controlled data on some participants. The fact that prodromal patients in LipiDiDiet reaped greater benefits than did mild AD patients in earlier trials suggests that Souvenaid is likely to have a better effect the earlier it is given, Hartmann said. Clinicaltrials.gov lists a planned Phase 2 trial that will test the effects of one year of Souvenaid on cognitive aging in 120 cognitively healthy elderly participants.
Calming Astrocytes Aids Huntington’s Brain
Inflammation is a major driver of pathogenesis in Alzheimer’s and other neurodegenerative diseases, and many groups are looking for ways to tweak this complex process. For example, the biotech company Vaccinex in Rochester, New York, is targeting the pro-inflammatory factor semaphorin 4D. First discovered as an axon guidance molecule in the developing nervous system, semaphorin 4D is now known to be released from neurons in response to stress or injury in the adult brain, said Elizabeth Evans of Vaccinex. Secreted Sema4D binds to receptors on glial cells, triggering secretion of inflammatory cytokines and collapse of their cytoskeletons. The net effect is to activate microglia and astrocytes, inhibit myelination, and disrupt the blood-brain barrier (Smith et al., 2015).
Evans noted that, once activated, astrocytes retract their processes from synapses. They no longer mop up excess glutamate from the synaptic cleft, which can lead to excitotoxicity and kill those synapses. Activated astrocytes also pull back from blood vessels, and so cannot take up glucose from the bloodstream to feed to neurons.
The researchers generated an antibody, pepinemab (previously known as VX15), that binds Sema4D and blocks its signaling (Fisher et al., 2016). In the YAC128 mouse model of Huntington’s disease, treatment with pepinemab slowed brain atrophy and improved some behaviors (Southwell et al., 2015).
At AAT-AD/PD, Evans discussed data from a second HD mouse model, Q175, as well as from a clinical trial. Immunostainings of Q175 brains revealed that neurons increasingly turned up Sema4D expression as disease progressed, starting at three months, before symptoms appeared. Adding Sema4D to isolated rat astrocyte cultures caused the cells’ actin cytoskeleton to collapse. They became pro-inflammatory, confirming an effect of Sema4D on these cells.
In sections from human postmortem HD brains, Sema4D was likewise elevated over controls, correlating with disease stage and with neuronal death in multiple cortical regions. Immunostainings of these brain sections showed that, in the presence of high Sema4D expression, astrocytes pulled back their processes and balled up. Together, the data suggested that blocking Sema4D might help preserve normal astrocytic functions and brain glucose metabolism.
To test this, the researchers initiated a Phase 1/2 trial, SIGNAL-HD, of pepinemab in 36 people with the causative CAG expansion. All were in either the late prodromal or early manifest stage of the disease. Half the cohort received a monthly infusion of 20 mg/kg pepinemab for six months, the remainder placebo. This trial met its primary endpoint of safety and tolerability, Evans noted.
A secondary endpoint, FDG PET, measured brain glucose metabolism. A preliminary interim analysis of 11 people on pepinemab and eight people on placebo found consistent differences between the groups. In controls, the FDG signal fell over the course of the study, while in the treatment group, it rose. This was consistent across the whole cohort. It occurred in every brain region, with the largest effects seen in frontal and parietal cortex.
After six months, the placebo group “crossed over” to receive pepinemab. Six months later, their FDG PET signals had risen to the level seen in the treatment group. “We were encouraged by the magnitude and consistency of the change in FDG PET signal in two different cohorts of patients,” Evans said. She said they also saw encouraging trends on brain atrophy, motor skills, and cognition, although she did not show those data.
Based on these findings, Vaccinex has launched a larger Phase 2 study, enrolling 179 people with early manifest HD and 86 with late prodromal disease. The former will receive 18 months of treatment with 20 mg/kg pepinemab, the latter 18 or 36 months. This study is fully enrolled, with the last patient visit scheduled for July 2020 and topline data expected in October. Evans did not say whether the current coronavirus pandemic would affect this timeline.
The researchers will also test pepinemab in AD patients. In postmortem sections from AD brains, Sema4D rises with disease progression just as it does in HD. The Phase 1b trial will enroll 60 people with MCI due to AD or mild AD. Twenty of them will receive monthly infusions of placebo, 20 will get 20 mg/kg pepinemab, and 20 will get 40 mg/kg for 32 weeks. The primary objective is safety and tolerability, with FDG PET as a secondary outcome. Other secondary and exploratory outcomes include cognitive measures such as the ADASCog and CDR, MRI volumetric scans, and fluid biomarkers of Aβ, p-tau, NfL, and cytokines. The trial is slated to start in July.
Epigenetic Drug Nudges Biomarkers of Inflammation and Degeneration
Next up, a drug that targets epigenetic regulation. Researchers at Oryzon Genomics in Barcelona, Spain, developed vafidemstat, also known as ORY-2001, as a selective inhibitor of lysine-specific demethylase 1 (LSD1). This enzyme represses neuronal genes and is the most abundant histone demethylase in the frontal cortex. In unpublished work on animal models, inhibiting LSD1 with vafidemstat boosted learning and memory while lessening neuroinflammation and aggression. Vafidemstat also inhibits monoamine oxidase B (Fang et al., 2019).
Taken orally, vafidemstat enters blood and brain well, and appeared safe in Phase 1 trials (May 2017 conference news). At AAT-AD/PD, Oryzon’s Michael Ropacki elaborated on these data. He noted that more than 250 people have now taken the drug without ill effects. About 150 of those were on the drug for two months or more, while 40 people took it for more than a year.
Ropacki discussed interim data from the Phase 2a Epigenetic Therapy in Alzheimer’s Disease, or ETHERAL, trial. This one-year trial has a target enrollment of 150 participants with mild to moderate AD confirmed by biomarkers. Currently, it is fully enrolled in Spain, France, and the U.K., with a total of 117 participants from those locations. Enrollment in the U.S. is ongoing, with 21 participants from there so far. Ropacki showed six-month cerebrospinal fluid biomarker data from the European cohort, but cautioned that the analysis is ongoing and findings are preliminary.
The European participants are nearly all white, with an average age of 73. Fifty-five percent of them have mild AD, the remainder moderate. In this cohort, 38 people take 0.6 mg vafidemstat, 34 take 1.2 mg, and 45 take placebo. After six months, the inflammatory biomarker YKL40 dropped statistically significantly in CSF in those on drug. This appeared to be driven by an effect in people with moderate AD, rather than mild. CSF neurogranin, a marker for synaptic damage, trended lower in the treatment group, with the effect reaching significance in the moderate subgroup taking 0.6 mg vafidemstat. CSF NfL, a marker of neurodegeneration, rose in all participants as disease progressed, but its rise was significantly dented in the mild subgroup taking 1.2 mg vafidemstat. As expected from vafidemstat’s mechanism of action, there were no changes in cerebrospinal fluid biomarkers of Aβ and tau after six months. In addition, participants on drug did no better than the placebo group on the ADAS-Cog14. Ropacki noted this trial was not powered to show changes in cognition.
“ETHERAL has produced interesting biomarker data that require further exploration and replication in the larger combined European and U.S. samples,” Ropacki said. The drop in YKL40 is the first evidence in people of the anti-inflammatory effect seen in animal models, he added.
Because vafidemstat treated aggression and apathy in animal models, the researchers are also studying behavioral effects in people. In small trials of people with autism spectrum, borderline personality, and attention-deficit and hyperactivity disorder, the drug significantly calmed agitation and aggression, Ropacki said. This led the researchers to offer an open-label substudy of the ETHERAL trial, dubbed REIMAGINE AD, to examine behavioral change in AD. It enrolled 12 AD patients from a single site in Barcelona. On several measures of aggression, namely the Cohen-Mansfield Agitation Inventory, Neuropsychiatric Inventory Agitation/Aggression subscale, and the Clinical Global Impression Improvement scale, participants improved markedly, with scores dropping by up to 80 percent from baseline. The findings were significant at the 0.05 level. On two caregiver measures, the Zarit Caregiver Burden Interview and NPI emotional distress scale, scores dropped similarly. However, the benefits took longer to show up in these AD patients than they had in cohorts of younger people, Ropacki noted.
Could Suppressing Cortisol Boost Cognition in AD?
Stress has also been implicated in AD, with evidence that high levels of glucocorticoids such as cortisol can worsen pathology (Dec 2011 news; Jun 2007 news). Cortisol has numerous harmful effects, including raising Aβ levels, boosting excitatory signaling and neurotoxicity, and dampening long-term potentiation. However, few groups have tried to target this mechanism in AD.
At AAT-AD/PD, Craig Ritchie of the University of Edinburgh described an approach being pursued by the biotechnology company Actinogen, based in Sydney. Ritchie noted that he is a paid consultant for Actinogen. The company developed a small molecule, Xanamem, that reversibly binds to the enzyme 11beta-HSD1 and inhibits it. Because this enzyme converts inactive cortisone to active cortisol, the inhibitor suppresses cortisol production (Webster et al., 2017).
In the Tg2576 mouse model of amyloidosis, nine to 12 months of Xanamem lowered cortical plaque load and improved memory in the Morris water maze. When started early, it prevented cognitive decline (Sooy et al., 2015). In healthy or diabetic old people, a different 11beta-HSD1 inhibitor was shown to improve verbal fluency or memory, respectively, after four or six weeks of treatment (Sandeep et al., 2004).
Based on these data, the researchers took Xanamem into human trials. In the XanADu Phase 2 study, they gave 10 mg Xanamem daily for 12 weeks to 186 people with mild AD at 25 sites in Australia, the U.S., and the U.K. This did not affect cognition, but did alter levels of stress hormones, including boosting the inactive form, cortisone, as expected. Analysis of these data suggested the dose had been too low, so the researchers doubled it. In the XanaHES Phase 1 study in Perth, Australia, 30 healthy old people took 20 mg daily, while 12 took placebo for 12 weeks. After four weeks, participants on drug did better than the placebo group in CogState tests of working memory, visual attention, and psychomotor function. These improvements were sustained for the rest of the study, and had large effect sizes, with Cohen’s ds of around 0.6 to 0.8, Ritchie noted.
Because 20 mg Xanamem appeared to benefit cognition in healthy aging, but 10 mg did not help cognition in mild AD, the researchers decided to next test 20 mg in prodromal AD. They are also planning trials in schizophrenia and Type 2 diabetes, conditions marked by high cortisol and cognitive impairment. “11beta-HSD1 inhibition with Xanamem is a targetable and rational approach to therapy in AD, with a range of additional potential therapeutic applications,” Ritchie noted.
Will Making Synapses More Plastic Sharpen Memory in HD?
Synapses falter in Alzheimer’s and other neurodegenerative diseases. Could propping them up slow decline? Aaron Koenig of Sage Therapeutics in Boston described SAGE-718, a derivative of the endogenous steroid 24(S)-hydroxycholesterol. SAGE-718 binds to the NMDA receptor and enhances its function. The steroid does not directly activate the receptor, but acts as a positive allosteric modulator, or PAM. NMDA receptor activity induces long-term potentiation of synapses, and thus is essential for learning and memory.
Notably, in the observational TRACK-HD study, which follows biomarker and clinical change in Huntington’s patients, plasma 24(S)-hydroxycholesterol levels waned as symptoms appeared, correlating with worse performance on tests of executive function and emotional processing (ACNP poster presentation M-147; Tabrizi et al., 2012). An older study also reported a drop in 24(S)-hydroxycholesterol in symptomatic HD (Leoni et al., 2008). The findings suggested SAGE-718 might help HD patients.
The researchers first tested SAGE-718 in several small Phase 1 studies of healthy volunteers, who took either the steroid or a placebo pill, and then a low dose of ketamine to inactivate their NMDA receptors. Electrophysiology and fMRI measures showed that SAGE-718 counteracted ketamine, normalizing synaptic activity. In the largest study, 19 participants took SAGE-718 for 11 days, 21 placebo. Every other day, they took CogState card-playing tests designed to evaluate working memory, attention, spatial learning, and executive function. On the simpler tasks, treatment and placebo groups performed equally well, but a difference emerged on more demanding tasks. In the “two back” card test of working memory, where participants need to recall the card they saw two card flips ago, those taking SAGE-718 outperformed the placebo group at every time point. On the Groton maze learning test of executive function, participants on SAGE-718 did better starting at day six. They also trended toward better performance on a test of spatial learning.
Based on these results, the researchers launched a pilot study of SAGE-718 in Huntington’s disease, which is characterized by executive dysfunction in addition to the signature motor problems. Six people with mild to moderate HD took SAGE-718 every other day for two weeks, with repeated testing on the Cogstate tasks. After one week, their performance on the “two back” card test improved, and remained high for the rest of the study. Koenig noted that the magnitude of improvement was greater than that seen in healthy controls in the previous study. There was no control group in this study.
“We believe this early signal holds promise for showing effects that would ultimately be meaningful for patients with Huntington’s-related cognitive impairment,” Koenig said. Further investigation of SAGE-718 is warranted for conditions associated with NMDA hypofunction, including cognitive deficits in early HD, he added.—Madolyn Bowman Rogers
Fisher TL, Reilly CA, Winter LA, Pandina T, Jonason A, Scrivens M, Balch L, Bussler H, Torno S, Seils J, Mueller L, Huang H, Klimatcheva E, Howell A, Kirk R, Evans E, Paris M, Leonard JE, Smith ES, Zauderer M.
Generation and preclinical characterization of an antibody specific for SEMA4D.
MAbs. 2016;8(1):150-62. Epub 2015 Oct 2
PubMed.
Does Alzheimer’s Start in the Heart of the Cholinergic System?
Three the approved drugs for Alzheimer’s disease attempt to boost the brain’s supply of acetylcholine, a critical neurotransmitter for cognition that wanes in people with the disease. But might crumbling of the brain’s cholinergic system be one of the earliest changes in the preclinical stages of the AD cascade? According to new evidence from longitudinal studies presented at the virtual AAT-AD/PD meeting, held remotely April 2 to 5, the answer could be yes. Buoyed by advances in brain mapping and neuroimaging, researchers reported that in cognitively normal people with AD biomarkers, shrinkage of the nuclear basalis of Meynert (NbM)—an already tiny speck in the basal forebrain—predicted a rise in markers of neuroinflammation, shrinkage in the medial temporal lobe, and cognitive decline. Damage to the diminutive NbM also foreshadowed dementia in people with Parkinson’s disease. Together, the findings bestow a minuscule patch of cells deep in the brain with outsize power—or vulnerability—in neurodegenerative disease.
Cholinergic neurons reside in distinct clusters, called nuclei, within the basal forebrain and within the brainstem. Projections from these nuclei supply acetylcholine (ACh) to different regions of the brain. The NbM, aka Ch4, is the major cholinergic supplier of the cortex, and is more strongly tied to cognition than any of the other nuclei. Scientists visualize cholinergic neurons in postmortem samples by immunostaining for the enzymes choline acetyltransferase (ChAT) and acetylcholine esterase (AChE). In recent years, advanced imaging and stereology have created precise anatomical maps of the NbM, facilitating longitudinal measurements of its size via structural MRI (Zaborsky et al., 2008; Kilimann et al., 2014).
Applying these techniques in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort, researchers led by Taylor Schmitz and Nathan Spreng previously tied smaller NbM volume to subsequent shrinkage of the entorhinal cortex (EC) and to cognitive decline (Nov 2016 news). They placed degeneration in the NbM upstream of atrophy in the EC, to some controversy in the field.
At AD/PD, Sara Fernández-Cabello of the University of Salzburg in Austria presented follow-up ADNI data, which included more participants, more timepoints, and more powerful statistical analyses connecting the dots between degeneration of the NbM and other regions of the brain. Drawing on more than 835 participants in ADNI who had CSF measurements and structural MRI scans over two years, Fernández-Cabello reported that small NbM size at baseline predicted subsequent shrinkage of the EC, but not vice versa. In turn, a smaller EC predicted atrophy in temporal cortical regions including the entorhinal, perirhinal, parahippocampal, and fusiform cortices and temporal poles, as well as in some clusters within the insular and posterior parietal cortices.
Importantly, this degenerative chain of events took place only in people with an abnormal CSF ratio of p-tau to Aβ42, regardless of their cognitive status. In other words, they represent steps of AD-specific pathogenesis. The findings were published March 1 in Brain (Fernandez-Cabello et al., 2020).
NbM Upstream? An updated version of the AD neurodegenerative staging scheme places the NbM upstream of the EC. [Courtesy of Fernandez-Cabello et al., Brain, 2020.]
At AAT-AD/PD, Fernández-Cabello showed additional, unpublished findings from the same ADNI cohorts, linking degeneration in the NbM with different types of cognitive dysfunction. The researchers found that a smaller NbM at baseline predicted worsening scores on tests of memory and executive function. They then pieced together which change mediated which other change. They found that, among people with abnormal AD biomarkers, the memory loss triggered by NbM degeneration was mediated via atrophy in temporoparietal and medial regions. In all, the findings suggest a pathological chain of events unfolding uniquely in people with AD biomarkers, in which shrinkage of the NbM leads to subsequent degeneration of temporoparietal regions, ultimately manifesting in memory loss.
At AD/PD, Taylor Schmitz of Western University in Ontario described another consequence of NbM degeneration in people with AD: the transformation of microglia from helper to harmer. Numerous studies have described how microglia transition from a homeostatic, or neuroprotective, state, to an inflammatory, neurotoxic state in people with AD and in animal models of amyloidosis and tauopathy (Jan 2017 news; Sep 2017 news; May 2019 news). Schmitz blames a floundering cholinergic system, citing previous findings that cholinergic stimulation not only quells inflammation in the peripheral nervous system, but also calms microglia in the CNS via stimulation of microglial alpha 7 nicotinic acetylcholinergic receptors (nAChR) (Wang et al., 2003; Shytle et al., 2004; De Simone et al., 2005) Does a dwindling cholinergic supply unleash toxic microglial inflammation?
To test this idea, Schmitz used longitudinal imaging and biomarker data from ADNI. He partitioned 268 cognitively normal volunteers into “neurotypical aging” or “preclinical AD” subgroups based on their CSF p-tau/Aβ42 ratio. He then tracked NbM volume on MRI over two years, finding that people with abnormal biomarkers not only had a smaller NbM at baseline, but also lost more NbM volume than people in the neurotypical aging group did. The researchers also tracked soluble CSF TREM2, as its rise coincides with the emergence of tau pathology in AD and is thought to signify a transition of microglia into a neurotoxic state (Jan 2016 news).
Schmitz and colleagues found that typical aging and preclinical AD groups started out with similar concentrations of sTREM2. Over two years, sTREM2 rose slightly in both groups, with the latter trending toward more elevation than the former. NbM shrinkage and rising sTREM2 concentration were strongly correlated, but only in the preclinical AD group.
Mirroring the CSF sTREM2 finding, plasma complement C3 also crept up along with NbM degeneration in the preclinical AD group. Together, the findings support the idea that in preclinical AD, NbM degeneration correlates with a rise in neuroinflammation. The results were published February 26 in the Journal of Neuroscience (Schmitz et al., 2020).
Schmitz acknowledged that he cannot formally prove that NbM degeneration causes neuroinflammation in preclinical AD. Nor does he know what drives C3 to increase in the blood in preclinical AD. This could derive from signals in the brain, for example cytokines that inflame the vascular endothelium of the blood brain barrier. Alternatively, a loss of cholinergic output from the vagus nerve could lead to activation of macrophages throughout the body. It was via this peripheral pathway that the anti-inflammatory effects of acetylcholine were first established, Schmitz noted.
Another limitation of the ADNI volume data is its lack of specificity for cholinergic neurons, Schmitz said. Theoretically, dying non-cholinergic cells in close proximity to cholinergic neurons could contribute to shrinkage in the NbM. Schmitz and colleagues are developing a PET tracer for vesicular acetylcholine transferase, a protein expressed in presynaptic terminals of cholinergic neurons. Mouse experiments show a cholinergic system all aglow, Schmitz said, and a pilot study testing the tracer in humans is underway.
Researchers led by Zhude Tu at Washington University in St. Louis have developed another VAChT PET tracer. In his abstract at AD/PD, Tu reported that the first human study of that tracer showed cholinergic synapses in multiple regions of the brain decrease with age.
Yet another approach to gauge if the cholinergic system is healthy is to measure the integrity of the axonal projections that spring forth from the basal forebrain. At AD/PD, Milan Nemy of the Czech Technical University in Prague described how he used diffusion tensor imaging to trace cholinergic circuitry in 262 healthy volunteers between 39 to 77 years of age (Nemy et al., 2020). Nemy also took stock of the NbM volume and white-matter hypointensities, a proxy for small-vessel disease. Nemy reported that the cholinergic circuitry eroded with age; this erosion correlated more strongly with poorer performance on memory and attention tests than did white-matter hypointensities or NbM volume, suggesting that weakening cholinergic connections could manifest as cognitive decline.
Schmitz considers the white-matter tractography data compelling, in that it likely captured at least some of the basal forebrain projectome. Still, he believes, tractography comes with the same limitation as measuring NbM volume: It’s not specific to cell type. The farther one moves from the basal forebrain, the more difficult it is to definitively link axonal projections to cholinergic neurons, Schmitz said, suggesting that, ultimately, PET tracers will most clearly delineate the cholinergic system.
A puny NbM also foretold of cognitive trouble in people with Parkinson’s. In her presentation at AAT-AD/PD, Joana Pereira of the Karolinska Institute in Stockholm described findings from the Swedish BioFinder study, which tracked NbM volume and cognition over an average of four years in a cohort of 106 nondemented people with PD, 20 people with Parkinson’s Disease Dementia (PDD), and 42 healthy controls (Pereira et al., 2020).
Forebrain Foretells Dementia in PD? The nucleus basalis of Meynert (Ch4, green) is smaller in people with PDD or PD who later converted to dementia (PD-c) than in people who never developed dementia (CTR, PD-s). [Courtesy of Pereira et al., Neurobiology of Disease, 2020.]
At baseline, people with PDD, or those with PD whose cognition would later decline, had smaller NbM volumes than did healthy controls or people with PD who did not. NbM atrophy over time associated with waning global cognition and verbal fluency, as well as diagnosis of dementia. The medial septum/diagonal band—another cholinergic cluster in the basal forebrain—also shrank more over time in people with PDD or in those who later developed dementia. Together, the findings suggested that degeneration of cholinergic basal forebrain structures might influence the clinical heterogeneity of PD.
“There are now several studies showing that the NbM could be a non-invasive marker of cholinergic dysfunction and dementia in AD and PD,” Pereira wrote to Alzforum. Despite substantial overlap in NbM volume between groups, Pereira proposed that NbM atrophy could be used to identify people at highest risk for cognitive decline, or even as a biomarker in clinical trials aimed at preventing dementia.—Jessica Shugart
‘Working from Home’: Do Gut Microbes Hold Sway Over Glia, Aβ?
At the virtual AAT-AD/PD meeting, held April 2 to 5 online, researchers substantiated some prior hints on potential causal connections between the throngs of bacteria inhabiting the gut, how microglia function in the brain, and deposition of Aβ plaques. Using antibiotics, caloric restriction, stool transplants, or probiotics to shift the gut microbiome, scientists reported that they were able to modulate microglial phenotypes and Aβ burden. They zeroed in on specific bacterial strains that either helped or hindered the brain’s immune cells, and uncovered profound differences between male and female mice in this regard. As they begin to drill down into details of the elusive gut-brain connection, some researchers envision microbiome-based therapies for neurodegenerative disease.
Sangram Sisodia of the University of Chicago reported striking sex-specific effects of the microbiome on the brain. Previously, Sisodia’s group had found that treating APP/PS1 mice with broad-spectrum antibiotics did not destroy the microbiota in their gut, but altered their composition. This remodeling differed in males and females. Males had an increase in certain Bacteriodes species and a drop in a strain of E. coli; females saw more Akkermansia, E. coli, and Proteobacteria, while Bifidobacterium species plummeted.
Notably, these microbiome changes appeared to affect the brain in sex-specific ways (Dodiya et al., 2019). In antibiotic-treated APP/PS1 males, microglia took on a homeostatic gene expression signature, whereas untreated APP/PS1 microglia have the neurodegenerative disease phenotype (MGnD). Treated male mice also had a lower amyloid plaque burden. In contrast, antibiotic-mediated microbiome remodeling had no such effect on the brain in female mice.
At AD/PD, Sisodia showed some detail on causal connections between the microbiome, microglia, and amyloid plaques. He noticed that the benefits of antibiotic treatment in the male APP/PS1 mouse brain were wiped away when the animals subsequently received regular doses—via gastric gavage—of fecal matter from untreated mice. Their microglia rounded up, retracted their processes, and re-expressed the neurodegenerative gene signature, which included higher levels of TREM2 and ApoE4. And plaques roared back. Essentially, the fecal transplant reverted animals back to their pre-antibiotic disease state, establishing that the gut microbiota indeed modulated microglial phenotypes and Aβ burden. To confirm that microglia caused the changes to Aβ burden, Sisodia’s group wiped out the mice’s microglia using a CSF-1R antagonist, which deprives these cells of survival signals (Sep 2019 news). In these mice-sans-microglia, fecal transplants did not affect Aβ plaque burden.
In all, Sisodia’s data suggested that the composition of microbes in the male gut influences form and function of microglia in the brain, which, in turn, affects Aβ deposition.
The reason behind this sex difference in microglial response is unclear, Sisodia said. It may lie beyond microbiome remodeling. Other studies have described sex-specific microglial phenotypes, as well as differences in the way the microbiome shapes microglial development in the two sexes (Thion et al., 2018; Jun 2018 news; Jul 2019 conference news).
What exactly are the gut microbes that hold sway over microglia and Aβ plaque deposition in a mouse’s brain? At AD/PD, Howard Weiner of Brigham and Women’s Hospital in Boston had some tentative answers. Previously, this group had reported that Tg2576 mice on a calorie-restricted diet had fewer Aβ plaques. The meager chow reduced numbers of Bacteriodes species, while boosting Faecalibaculum (Cox et al., 2019).
The researchers reported that feeding APP/PS1 mice on normal chow with Bacteriodes fragilis upped amyloid plaque load. Bacteriodes have been independently reported to rise in the human gut with age and AD (Odamaki et al., 2016; Vogt et al., 2017).
At AD/PD, Weiner reported that supplementing the chow of mice with a strain of Faecalibaculum had the opposite effect on microglia. Feeding this bacteria to mice converted their microglia from an MGnD phenotype to a homeostatic state, and lessened expression of inflammatory cytokines in their brains. Interestingly, Weiner reported that in the cortices of mice fed Bacteriodes fragilis, expression of three genes known to promote amyloidogenic processing of APP—Cdk5, Gga1, and Prkcb—shot up. Together, the data suggested that specific bacterial species in the gut could modulate Aβ deposition not only by influencing microglia, but also by tweaking Aβ42 production.
Finally, Weiner reported that treating APP/PS1 mice with metronidazole, an antibiotic that targets anaerobic bacteria including Bacteroides, lessened Aβ plaque load. The effect was stronger in female mice, which started out with a higher plaque load than males. On the face of it, this result contrasted with Sisodia’s. The cocktail of five antibiotics Sisodia’s group used included metronidazole and benefited males more than females. Sisodia told Alzforum that, according to data in an upcoming paper from his lab, treatment with metronidazole, or any other lone antibiotic, did not affect plaque load.
Maria Doitsidou of the University of Edinburgh used a reductionist approach to identify a bacterial strain that assuaged a different neurodegenerative proteopathy, that is, α-synuclein aggregation. Doitsidou reviewed recent data from a C. elegans model of synucleinopathy (Goya et al., 2020). In a nutshell, the researchers screened 40 bacterial strains for their ability to prevent α-synuclein aggregation in worms broadly overexpressing a fluorescently tagged version of the human protein in muscle. The probiotic B. subtilis strain PXN21 did the trick. While untreated worms were full of fluorescent synuclein aggregates, those that noshed on B. subtilis had none. What’s more, switching the worms from their normal diet of E. coli to the probiotic strain later in life dissipated aggregates that had already formed.
The findings independently confirm previous C. elegans studies led by Roberto Grau at Rosario National University, Argentina. He had found that B. subtilis blocked α-synuclein aggregation and delayed neurodegeneration (Apr 2018 conference news). Grau’s study appeared in the February issue of Journal of Alzheimer’s disease (Cogliati et al., 2020).
For her part, Doitsidou took stock of gene-expression changes in C. elegans in response to eating B. subtilis versus E. coli. Among the genes expressed differently were those involved in lipid metabolism, specifically of sphingolipids. Several genes pegged as PD risk factors are involved in sphingolipid processing, including GBA1. Other groups have linked microbial short-chain lipids to microglia and Parkinson’s (Dec 2016 news).
Doitsidou said that her lab is testing B. subtilis extracts on PD patient-derived dopaminergic neurons, and feeding the probiotic strains to mouse models of synucleinopathy. She hopes to identify the specific bacterial metabolites that quash α-synuclein aggregation. In addition, the researchers are working with Edinburgh neurologists to start a clinical trial that will evaluate B. subtilis probiotics in people with PD.
Giovanni Frisoni of the University of Geneva presented a nugget of human data on this topic. People with AD, PD, and other neurological disorders have altered gut microbiomes, and mouse fecal transplant studies suggest that these shifted microbiomes could cause or exacerbate disease (Cattaneo et al., 2017; Dec 2016 news; Feb 2017 news). Frisoni wondered whether, on the flip side, fecal transplants from people without AD, especially people at lowest risk for the disease, might protect against it.
To test this idea, researchers dosed 1-year-old 3xTg mice with fecal matter from one of four people at varying risk for Alzheimer’s. One 76-year-old donor with an ApoE3/4 genotype already had the disease. The three other donors were cognitively normal: a 65-year-old with one ApoE4 allele, a 72-year-old ApoE2 carrier, and a 24-year-old ApoE3 homozygote. Frisoni reported that 3xTg mice that received stool via a feeding tube from the person with AD displayed more AD-like behavior, including more anxiety and less movement, than those who received stool from other 3xTg mice, or saline. Mice gavaged with stool from the young donor or from the ApoE2 carrier performed better on a novel object recognition test of memory than did mice treated with stools from any of the other donors, or saline. Stool from the young donor or the ApoE2 carrier also appeared to reduce plaque load in the 3xTg mice. Frisoni stressed that the experiments were preliminary, and part of ongoing analyses.
The mechanism underlying these effects is unclear. Still, they imply that medications processed from feces of healthy people might, in theory, somehow stave off disease in those who are at risk.—Jessica Shugart
Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, Blecher R, Ulas T, Squarzoni P, Hoeffel G, Coulpier F, Siopi E, David FS, Scholz C, Shihui F, Lum J, Amoyo AA, Larbi A, Poidinger M, Buttgereit A, Lledo PM, Greter M, Chan JK, Amit I, Beyer M, Schultze JL, Schlitzer A, Pettersson S, Ginhoux F, Garel S.
Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner.
Cell. 2018 Jan 25;172(3):500-516.e16. Epub 2017 Dec 21
PubMed.
Aβ in Lewy Body Disease: Two Diseases at Once, or Another Beast Entirely?
Many people with Lewy body diseases (LBDs) such as Parkinson’s ultimately develop dementia, and many have Aβ plaques and tau tangles. Do they have two diseases at the same time, or is this combination of scourges a unique entity unto itself? At the virtual AAT-AD/PD meeting, held April 2 to 5, researchers reported that people with PD who carry genetic risk variants for Alzheimer’s disease were more likely to become cognitively impaired. Similarly, AD variants predicted which PD patients harbored Aβ and tau proteopathies in their brains. In people with dementia with Lewy bodies (DLB), Aβ plaques seemed to worsen cognitive decline more than tau tangles did, suggesting an etiology distinct from AD. Although the interactions between α-synuclein, Aβ, and tau still need more clarification, some suggested that therapies targeting Aβ might benefit people with Lewy body diseases.
LBDs include PD, PD dementia (PDD), and DLB. While cognitive symptoms arise prior to movement problems in people with DLB, the opposite is true for PDD. More than 80 percent of people with PD ultimately develop dementia, but cognitive trajectories vary widely between patients. Age, male sex, severity of motor symptoms, and sleep disorders raise a person’s risk of PDD, and CSF biomarkers of AD, as well as ApoE4 genotype, have been associated with cognitive decline in PD (Tropea et al., 2018; Feb 2020 news).
Might other AD variants also influence cognitive worsening in PD? At AD/PD, Thomas Tropea of the University of Pennsylvania in Philadelphia addressed this question using genetic and longitudinal cognitive data from 305 participants in UPenn’s ongoing PD cohort. The volunteers had PD but were dementia-free when they enrolled. They took the Mattis Dementia Rating Scale-2—a cognitive composite—every year for an average of 4.4 years and up to 12 years. Tropea, who is part of Alice Chen-Plotkin’s group at UPenn, screened the participants for 22 genetic risk variants, including 20 confirmed and two candidate variants from AD GWAS (Mar 2019 news).
By now, half of the participants have dementia, and Tropea reported that certain gene variants correlated with their rate of decline on the DRS-2. While ApoE4 had the strongest effect, AD risk variants in BIN1, NYAP1, PICALM, FERMT2, SLC24A4, and ADAM10 also associated with a faster downturn. Only PICALM and ApoE4 associated with decline on all five cognitive domains included in the DRS-2; other variants correlated only with particular components. Tropea next wants to investigate the combined effects of these AD risk variants in predicting cognitive decline in people with PD.
Do AD genes also flag AD neuropathology in people with PD? Also at AAT-AD/PD, David Dai in Chen-Plotkin’s group drew on genetic and neuropathological data from 208 people who died with an LBD—55 with PD, 108 with PDD, 45 with DLB. Based on their burden of Aβ plaques and tau tangles at autopsy, Dai ranked the degree of AD neuropathology for each case as none, low, intermediate, or high. A third of the cohort had some degree of these proteopathies. Dai then fed genotypes at 20 AD risk loci from 127 of the patients into an algorithm that returned a combination of four factors—ApoE4, BIN1, and SORL1 genotypes, and age at onset—as best correlated. Dai combined these four into a risk score, which predicted the presence of plaques and tangles in a test cohort of 81 other cases. Breaking up risk scores into quintiles, Dai found that cases with risk scores in the highest quintile were four times more likely to have Aβ plaques and tangles than those in the lowest two quintiles.
All three genetic variants included in the score are implicated in Aβ production. SORL1 was more strongly associated with plaques and tangles in the LBD cohort, Dai reported. He plans to investigate this relationship further in larger cohorts, and to extend his studies to living PD patients with abnormal AD biomarkers.
How do interactions between Aβ, tau, and α-synuclein influence the clinical symptoms of Lewy body disease? Daniel Ferreira of the Karolinska Institute in Stockholm reported new results from a collaboration between European DLB consortia and the Mayo Clinic DLB cohorts (Jul 2019 news). Representing 11 centers and 417 participants, the consortia track biomarkers, cognition, and clinical progression in people with DLB. The European centers used CSF Aβ42 and p-tau as AD markers; Mayo, PiB and tau PET. They collaborated to generate cut-off values for each marker.
AD Markers in DLB. Nearly half of people with DLB had amyloid (A), with or without tau tangles (T). [Courtesy of Daniel Ferreira, Karolinska Institute.]
Ferreira reported that 39 percent of the trans-Atlantic cohort had normal Aβ and tau, while 32 percent had evidence of plaques, and 15 percent had both plaques and tangles. Interestingly, 13 percent were abnormal for tau but not Aβ. Ferreira noted that this could represent a type of tau accumulation distinct from the AD cascade.
Age most strongly predicted AD biomarkers: While 60 percent of the youngest DLB patients had neither amyloid nor tau pathology, only 20 percent of the oldest patients were negative for both. The proportion of people with abnormal tau, but not Aβ, held steady across age groups. Finally, in line with many prior studies, ApoE4 carriers in this cohort, too, were more likely to have plaques at a younger age than noncarriers.
How did these combinations of proteopathy influence DLB symptoms? Ferreira reported that while both Aβ and tau came with lower scores on the MMSE, Aβ influenced cognition the most. Tau did not associate with cognitive impairment independently of Aβ.
Curiously, the presence of tau pathology correlated with a lower risk for noncognitive symptoms of DLB, including parkinsonism and sleep disorders. The reason behind the apparent protective influence of tau tangles against certain DLB symptoms is unclear. Ferreira said this could likely complicate diagnosis. Strikingly, Aβ and tau did not synergize to influence cognition in DLB, as they do in Alzheimer’s. Together, the findings suggested that in people with DLB, Aβ is a stronger driver of cognitive decline than tau is.
Aβ Eggs on Cognitive Decline. Lewy bodies (top) drive cognitive impairment and other clinical features of DLB. Additionally, Aβ burden (A) contributes significantly to cognitive impairment (left), while tau tangles associate with a lower frequency of other clinical features of the disease (right). [Courtesy of Daniel Ferreira, Karolinska Institute.]
Ferreira concluded that while amyloid and tau pathology are both common in people with DLB, they have distinct influences on cognition. They appear to work differently than they do in people with AD, where Aβ instigates tau pathology, which associates strongly with cognitive decline. Future studies will examine how Aβ and tau influence specific cognitive domains in people with DLB, Ferreira said.
In his talk at AAT-AD/PD, John Growdon of Massachusetts General Hospital in Charlestown made the case that Aβ-targeted therapies might be able to stem cognitive decline in people with LBD. He reviewed a decade’s worth of evidence linking Aβ to cognitive decline and markers of neurodegeneration in people with synucleinopathies. Initially, Growdon and his colleague Stephen Gomperts had reported that Aβ deposition predicted cognitive decline over a five-year follow-up in people with PD, and sped up cognitive decline in people with PDD or DLB (Gomperts et al., 2013). In 2016, they added that neurofibrillary tangles—as per tau-PET—also correlated with worse cognition in people with LBD (Sep 2016 news). More recent neuropathological findings showed that when present, Aβ deposition correlated with the severity of neurofibrillary tangles, as well as Lewy body inclusions (Shirvan et al., 2019). Finally, Growdon said that Aβ deposition correlated with cortical thinning in people with LBD, which, in turn, associated with cognitive decline (Ye et al., 2020).
Together, the studies suggest that Aβ could exacerbate both tau and α-synuclein aggregation in LBD. This, Growdon believes, is why Aβ-targeted therapies could benefit people with synucleinopathies. There was no question-and-answer period at this virtual meeting, but Growdon anticipated the main question people might ask: Anti-Aβ therapies have failed to benefit cognition in people with AD, so why would they work for LBD? In LBDs, Aβ appears to exacerbate both tau tangles and α-synuclein aggregation, either of which could lead to dementia, Growdon said. Targeting Aβ could dismantle this toxic triplet, he said.
Second, while Aβ plays the role of instigator in AD, it appears to drive progression throughout the disease process in LBD. Therefore, while ridding the brain of Aβ may be too little, too late, for people with symptomatic AD, it could slow the disease in people with LBD, Growdon reasoned.—Jessica Shugart
Certain variants in the TREM2 gene more than triple a person’s risk of AD, seemingly by sapping the protective function this microglial receptor performs. According to findings presented at the virtual AAT-AD/PD meeting, held April 2 to 5, TREM2’s protective power falters as amyloidosis kicks into high gear.
Guojun Bu of the Mayo Clinic in Jacksonville, Florida, described what happened in a mouse model of amyloidosis when he switched on expression of human TREM2 at different stages of disease. Wild-type human TREM2 stemmed Aβ deposition, but only while plaques were in their infancy. It had no effect later on. In contrast, the R47H mutant never protected against plaques, and even exacerbated their growth. Together, the findings suggest that TREM2’s protective role is limited to early stages of Aβ deposition.
The heightened AD risk posed by rare variants in TREM2 point to a beneficial role for the wild-type variant, yet animal models have painted a complex and sometimes contradictory picture of the receptor’s influence on AD pathogenesis. For one, expression of TREM2 on microglia ramps up as amyloidosis worsens, yet microglia ultimately morph into ineffective, or even harmful, pro-inflammatory cells (Jan 2017 news; Sep 2017 news; Jun 2018 conference news).
Soluble TREM2, which is cleaved from the microglial surface, helps recruit microglia to plaques, yet levels of the snipped receptor rise in human cerebrospinal fluid as cognitive symptoms emerge in people with AD (Apr 2019 news; Jan 2016 news).
Flipping the TREM2 Switch. Tamoxifen induced expression of human TREM2 in 5xFAD mice at different stages of amyloidosis. [Courtesy of Guojun Bu, Mayo Clinic.]
How does TREM2’s role change throughout the long, drawn-out stages of amyloidosis? To address this question, Bu developed inducible human TREM2 mouse strains. Treating the mice with tamoxifen switched on expression of either human wild-type TREM2 or the R47H variant in microglia, while endogenous mouse TREM2 expression stayed constant. The researchers then crossed these TREM2 mice to the 5xFAD mouse model of amyloidosis. In the crosses, they induced human TREM2 expression during three phases of amyloidosis: early seeding and growth (birth to 3.5 months), rapid growth (2 to 5 months), or maturation (5 to 8 months) (see above).
When expressed from birth to 3.5 months, human wild-type TREM2 strongly protected against plaque formation. Mice expressing human wild-type TREM2 had about fourfold fewer plaques than did non-induced controls. In contrast, expression of R47H-TREM2 had no effect on plaque deposition at this early stage.
Everything changed when human TREM2 expression was on from 2 to 5 months of age. At this slightly older age, wild-type TREM2 expression no longer slowed amyloid deposition, and R47H made matters worse. Animals expressing the risk variant had two to three times more plaques than non-induced controls, though the plaques were about the same size in both strains.
The data suggest that during this phase of rapid plaque growth, wild-type TREM2 loses its protective function, while the R47H variant appears to exert a toxic function. In support of this idea, single-cell transcriptomic experiments revealed that the R47H mutant invoked more dramatic shifts in the microglial transcriptome than did the wild-type allele. In 5-month-old mice, R47H-TREM2 but not wild-type TREM2 upregulated genes involved in dysfunctional cell stress and energy metabolism pathways in microglia.
When expressed during the maturation phase of beta amyloidosis, from 5 to 8 months of age in these mice, neither wild-type nor mutant TREM2 affected Aβ deposition.
Together, the findings suggest that normal TREM2 thwarts amyloidosis early on, when plaques are just starting to develop. Once plaque growth takes off, TREM2’s protective function fades, and the detrimental effects of R47H become apparent. By the maturation phase, neither version of the human protein holds sway over amyloid.
The findings could have implications for the timing of therapeutic strategies aimed at revving up TREM2 signaling, Bu said. Such strategies would be most likely to work early in disease.—Jessica Shugart
At the second biannual Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held virtually April 2 to 5, companies offered some news on investigational Parkinson’s therapies in Phase 1. One approach is targeted toward a common form of PD caused by mutations in the glucocerebrosidase gene that slow down glycosphingolipid flux. Biotech company Lysosomal Therapeutics in Cambridge, Massachusetts, developed a small-molecule activator of glucocerebrosidase, LTI-291, that unclogged the recycling pathway and appeared to benefit brain function in Phase 1b. Taking a different tack, San Diego-based Neuropore Therapies deploys antagonists of the toll-like receptor 2 (TLR2) in hopes of dampening harmful immune responses while boosting autophagy of misfolded proteins. This approach not only is broadly applicable to all forms of PD, but also could help with proteinopathies beyond α-synucleinopathies. The company has decided to develop its Phase 1 compound NPT520-34 for amyotrophic lateral sclerosis.
Restoring Lipid Recycling
Up to 10 percent of Parkinson’s patients carry a mutation in the glucocerebrosidase (GBA) gene. These mutations heighten disease risk up to 15-fold, and cause a severe form of PD. GBA-PD progresses more rapidly than idiopathic disease and is more likely to lead to dementia. At AAT-AD/PD, Kees Been, who leads Lysosomal Therapeutics, discussed the company’s strategy for treating this form of PD. The glucocerebrosidase (GCase) enzyme catalyzes the breakdown of glucosylceramide to ceramide. This is a critical step in the recycling of membrane glycosphingolipids, which affect membrane properties. Mutations in GBA weaken GCase’s activity and slow the recycling. The researchers developed LTI-291, a small-molecule allosteric activator of GCase.
Controlling the Flow. GBA catalyzes a crucial step in the lysosomal breakdown of glycosphingolipids, allowing them to be synthesized anew and recycled to the plasma membrane. [Courtesy of Kees Been.]
In Phase 1 testing, about 1 percent of plasma LTI-291 entered the brain, achieving a concentration of 1 μM or better, based on the amount of free drug in CSF. This would be sufficient to double GCase activity, Been said. The compound has desirable drug properties, with a half-life of about 30 hours. It reached a steady state in the body after five or six days of dosing. About 98 percent of it became bound into membranes. This is necessary for it to work, since GCase resides in lysosomal membranes. That the fraction of free drug is small also helps reduce side effects, improving the compound’s safety, noted the company’s chief scientific officer, Peter Lansbury. “This drug is incredibly clean in toxicity assays. We have a huge therapeutic window,” Lansbury said.
Clearing the Backlog. In participants treated with glucocerebrosidase activator LTI-291, synthesis of downstream lipids (blue and red bars) surged seven and 14 days later, before stabilizing after one month. [Courtesy of Kees Been.]
In a Phase 1b trial, 40 people with GBA-PD took either placebo or 10, 30, or 60 mg/day LTI-291 for 28 days. In treated patients, the researchers measured a transient surge in glycosphingolipids (GSLs) downstream of ceramide, indicating an accelerating recycling rate. The surge subsided as lipid recycling stabilized, presumably at a higher level. Lansbury compared this effect to the temporary rise in water level when a dam is released, before the current reaches a new, faster, equilibrium. The increase in GSLs was greater in people with more severe GBA mutations, whose recycling rate had been more suppressed before treatment.
Improved Connectivity. Functional brain connectivity (yellow and red) in the default mode network improved (bottom) compared to baseline (top) in a Parkinson's patient taking LTI-291, getting closer to the activity seen in healthy controls (green). [Courtesy of Kees Been.]
The company also ran a Phase 1b imaging study of 14 people with GBA-PD. Participants took placebo, 10, or 60 mg/kg LTI-291 for 28 days. At the end of the study, brain function in treated participants had normalized on several measures. These included enhanced brain blood flow by arterial spin labeling, increased brain glucose metabolism by FDG PET, and better functional connectivity in the default mode network on fMRI scans. The effects were similar in all treated patients, but not statistically significant in this small cohort.
“We’re quite excited with these results,” Been said. Next, a Phase 2 study will test two doses plus placebo for one year in people with GBA-PD. The endpoints will be MDS-UPDRS score and cognitive change. Been said they are still deciding how many participants they need, but plan to power the trial to be able to detect a disease-modifying effect. He noted that this is the first allosteric activator of GCase to be tested in PD patients with a GBA1 mutation.
Dual Action on Inflammation and Autophagy
Another approach entering trials takes a different tack. At AAT-AD/PD, Douglas Bonhaus, who leads Neuropore Therapies, discussed the small molecule NPT520-34, an antagonist of TLR2. This immune receptor recognizes aggregated proteins and is expressed by both neurons and microglia. TLR2 expression rises as PD progresses, correlating with α-synuclein deposition (Dzamko et al., 2017).
In neurons, TLR2 turns down autophagy, which could contribute to the accumulation of misfolded proteins. In microglia, TLR2 signaling leads to activation of the NLRP3 inflammasome, which is associated with damaging responses to disease (Dec 2012 news; Nov 2019 news). Thus, TLR2 has a dual mechanism of action, the company’s Martin Gill, head of in vitro pharmacology, said at AAT-AD/PD.
The company identified NPT520-34 in a screen for compounds that boost autophagy. In cellular assays, the compound increased expression of the scaffolding protein LC3 about threefold, powering autophagy. In the L61 α-synucleinopathy mouse model, treatment with NPT520-34 dose-dependently lowered protein deposits. It also calmed neuroinflammation, with fewer activated microglia visible by TSPO imaging. NPT520-34 also preserved dopaminergic signaling and improved grip strength and walking, Gill said.
Notably, the compound stimulates clearance of other types of aggregate, as well. In the Line 41 mouse model of amyloidosis, it lowered amyloid plaques and TSPO signal. In SOD1-G93A mice, a model for amyotrophic lateral sclerosis, it curbed aggregates and inflammation in the spinal cord. When treatment began at symptom onset in these SOD1 mice, they survived for longer, 41 instead of 34 days.
In a first Phase 1 trial conducted in 2019 in 49 healthy volunteers, NPT520-34 appeared safe with repeated dosing of up to 500 mg per day, and had “well-behaved” pharmacokinetics. This maximum dose far exceeds the effective dose in animal models, equivalent to about 125 mg, Bonhaus noted. The most common adverse event was headache.
The company plans to evaluate this drug first in ALS, for which Neuropore Therapies received orphan drug designation in August 2019 (see press release). The researchers are currently doing a 28-day safety study in people with this disease. They hope to move forward with a three-month study that will include TSPO imaging, and then a six-month study of safety and efficacy.—Madolyn Bowman Rogers
α-Synuclein Antibody Misses Primary, May Have Signal on Secondaries
Roche and Prothena today announced topline results from the first half of their Phase 2 trial of the α-synuclein antibody prasinezumab. Results were negative on the MDS-UPDRS, the primary outcome measure, but the company said it saw signals of efficacy on multiple secondary and exploratory clinical endpoints. The findings were included in Roche’s quarterly earnings report, and no further details were available. Roche continues to analyze the data, and is discussing next steps with health authorities.
Meanwhile, at the second biannual Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held virtually April 2 to 5, researchers from Roche presented baseline data from this trial, as well as data suggesting that exploratory digital measures collected by smartphones may indeed be useful. Immunotherapy approaches remain popular, with AC Immune researchers at the conference describing an antibody candidate about to enter the clinic.
Because misfolded α-synuclein is believed to spread from cell to cell, numerous groups are targeting this extracellular form of the normally intraneuronal protein with antibodies. Biogen’s antibody BIIB054 is in Phase 2 but the company did not present at AAT-AD/PD (May 2018 conference news). Other candidates, such as AbbVie’s ABBV-0805 and AstraZeneca’s MEDI1341, remain in Phase 1.
Roche/Prothena’s prasinezumab is the furthest along clinically. It selectively recognizes aggregated α-synuclein over monomeric forms. Phase 1 data showed that it reached the brain (May 2017 conference news),so Roche launched the Phase 2 PASADENA study in 2017. This study consists of a one-year, placebo-controlled trial, followed by a one-year blinded extension where placebo participants will cross over to one of the two active doses (Apr 2018 conference news).
At AAT-AD/PD, Roche’s Gennaro Pagano reported baseline data from this trial. It enrolled 316 participants, two-thirds of them men, whose mean age was 60. They were fairly newly diagnosed with PD, having had the disease for an average of 10 months at baseline. A quarter were at Hoehn and Yahr stage 1, meaning they were mildly rigid, slow, or had a one-sided tremor. The remaining three-fourths were at stage 2, with symptoms on both sides of the body but no balance problems yet. Their mean score on the MDS-UPDRS was 31.4, indicative of early disease. PD patients average 68.4 on this scale (Goetz et al., 2008). The study is conducted at 64 sites in the U.S., France, Spain, Germany, and Austria.
This trial was designed using the Parkinson’s Progression Markers Initiative cohort as a reference. PPMI participants are also at an early stage, enrolling within three to six months of their diagnoses. However, PPMI participants are not allowed to be on monoamine oxidase-B inhibitors such as rasagiline or selegiline, while PASADENA participants are. The medication difference did not seem to affect the characteristics of this cohort, Pagano said. Adjusting for the difference in disease duration between the PPMI and PASADENA cohorts, the severity of motor symptoms was similar between the two groups, suggesting their disease progression rates may be similar too, Pagano said. He believes including patients on MAO-B therapy was the right call.
Roche’s previous ScarletRoAD trial of gantenerumab had disallowed concomitant AD therapies. This led to disproportionate dropouts in the placebo group, as people who noticed they were declining left the trial to go on those medications (Nov 2015 conference news).
The PASADENA cohort did differ from PPMI in other regards, with slightly less severe non-motor symptoms and a smaller dopaminergic deficit on DaT-SPECT imaging. Those aspects of disease might progress differently than predicted from the PPMI data, Pagano acknowledged. Overall, he concluded, “The PASADENA population can be considered representative of a wider PD population, and therefore suitable for testing the potential beneficial effects of drugs acting on disease progression.”
The trial’s primary outcome measure was change on the MDS-UPDRS total score, which evaluates motor and non-motor aspects of PD, over the first year of the study. Pagano said the researchers refined secondary outcome measures for this trial to take a more granular look at disease progression. They will separately evaluate scores from the MDS-UPDRS Part 1, which tracks non-motor symptoms; Part II, which measures movement-based activities of daily living; and Part III, which measures motor symptoms. They will also compare the time to worsening on each of these subscales, as well as take other clinical and cognitive measures such as the Clinical Global Impression Improvement, the Patient Global Impression of Change, the Schwab and England Activities of Daily Living Scale, and the Montreal Cognitive Assessment. The MDS-UPDRS were evaluated every eight weeks throughout the first year of the study, the other measures every six months. The study was designed to have 80 percent power to detect a 37.5 percent slowing of progression on the MDS-UPDRS over one year.
The topline results reported today apply to the placebo-controlled portion of the trial. The crossover portion is still running, although Prothena noted that some assessments have been missed due to the coronavirus pandemic.
The trial also includes exploratory digital measures of motor and non-motor abilities, evaluated by smartphone and a wearable wrist device (May 2017 conference news). Gait and mobility analyses are passively recorded. In addition, participants take tests of hand dexterity, speech, and balance, alternating tests on different days. Compliance tends to be a problem with mobile phone-based tests that require consistency, but Pagano said that preliminary data indicates that most participants are completing these tests five or six days every week.
Toward validation of those new tests, Roche’s Alessandra Thomann at AAT-AD/PD reported that digital measures of hand movement correlated with participants’ own assessments of their overall health and ability to function, and gait measures correlated with their ratings of their own mobility and discomfort. Roche’s Florian Lipsmeier reported that gait and arm movement measures dovetailed with MDS-UPDRS scores for slowness, rigidity, and activities of daily living, again suggesting these will be useful markers of progression.
A different therapeutic α-synuclein antibody is coming out of the labs at AC Immune in Lausanne, Switzerland. At AAT-AD/PD, AC Immune’s Elpida Tsika described their SupraAntigen method, which conjugates an antigen to a liposome that also contains adjuvants, to develop several antibodies that recognize different epitopes of α-synuclein. The researchers screened them to find the one with the best properties to take forward. The lead candidates were selective for aggregated over monomeric α-synuclein in dot blots, and had a 20 times slower dissociation rate from fibrils than monomers in surface plasmon resonance assays, indicating high affinity. In sections from postmortem PD brains, these antibodies labeled Lewy bodies.
Can they hold back pathology? When mixed with monomeric α-synuclein and aggregated seeds, the lead antibodies slowed in vitro fibril formation by 50 percent, from 30 to 45 hours. In a cellular assay, the antibodies dose-dependently lowered the amount of aggregates taken up by mouse primary cortical neurons, Tsika reported.
The researchers tested the three top candidates in M83 hemizygous transgenic mice expressing human A53T α-synuclein. They injected preformed α-synuclein fibrils into the anterior olfactory nuclei of 8-week-old mice, then treated them with 30 mg/kg antibody by intraperitoneal injection for four months. Antibodies ACI-mAb-1 and ACI-mAb-5 had the strongest effect, halving α-synuclein aggregates in contralateral brainstem and piriform cortex, and increasing body weight in treated animals. ACI-mAb-10 had little effect on pathology or body weight, but did preserve neurons in ipsilateral piriform cortex, as did ACI-mAb-1.
Tsika said a lead candidate was humanized last year and is in preclinical development now, but did not say which one was picked.—Madolyn Bowman Rogers

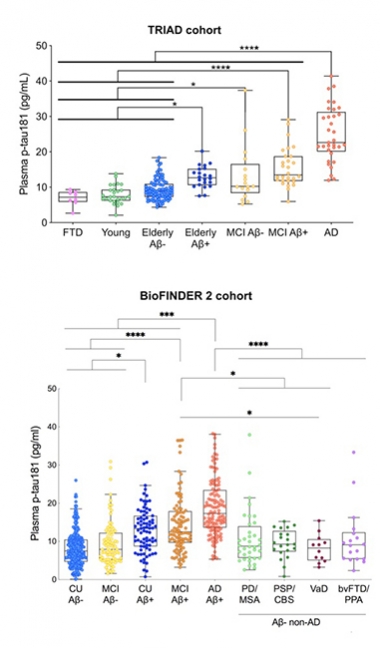


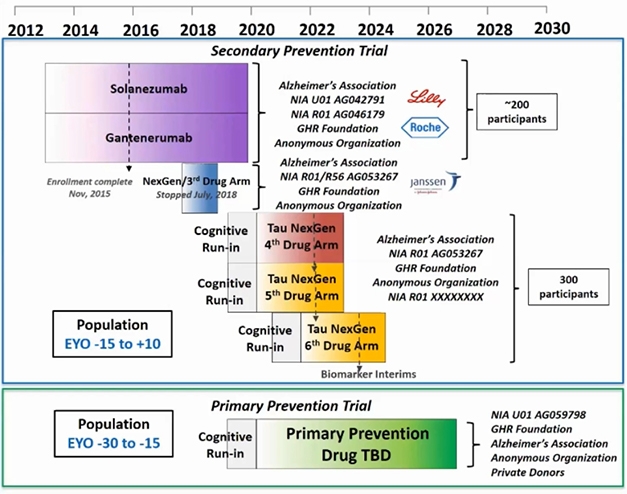



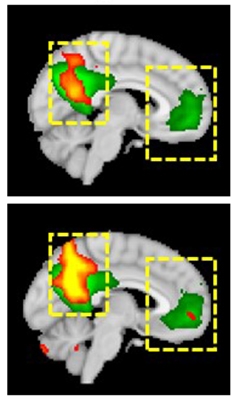
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.