Treating ALS in a Dish? Models Point to Drug Candidate for Sporadic Disease
Quick Links
Since sporadic neurodegenerative diseases have unknown and potentially multiple etiologies, cells derived from a single patient may not be the best option for screening drugs to treat all. Enter case clustering. As reported in the August 20 Nature Medicine, researchers led by Hideyuki Okano at Keio University School of Medicine in Tokyo have characterized the common patterns of degeneration in motor neurons from dozens of people with sporadic ALS. They then used these properties to find compounds that would stave off neurodegeneration. The dopamine receptor agonist ropinirole emerged as the top hit. It protected motor neurons derived from both sporadic and familial ALS patients.
- Researchers established a time frame of degeneration in induced motor neurons.
- Neurons derived from dozens of ALS patients used to screen known drugs.
- From 1,200 candidates, ropinirole emerged as most neuroprotective.
“The study is an incredible amount of work and a tour de force in using induced pluripotent cells to model ALS, including sporadic ALS,” commented Jared Sterneckert of the Technical University of Dresden.
The work also serves as an early validation for Answer ALS, said Jeffrey Rothstein of Johns Hopkins University in Baltimore. Rothstein directs the project, which aims to screen iPSC-derived motor neurons from more than 1,000 ALS patients and 100 controls to find new drugs. He said that while Okano’s findings are interesting, more samples would be needed, particularly control samples, to draw solid conclusions.
Though the vast majority of ALS cases are sporadic, researchers have relied extensively on models of familial ALS (FALS)—SOD1-ALS in particular—to study the disease. However, recent studies paint SOD1-ALS as a pathological outlier, characterized by distinct protein inclusions and a unique neurodegenerative cascade. Familial ALS caused by mutations in TDP-43 or FUS aligns more closely with sporadic disease, but models of sporadic disease itself are lacking (Kabashi et al., 2011; Da Cruz et al., 2017). An alternative is to study motor neurons derived from patients (Sep 2010 news; Apr 2013 news).
First author Koki Fujimori and colleagues took up this challenge. Before tackling sporadic ALS, the researchers grew motor neurons from induced pluripotent stem cells derived from four TDP-43-ALS cases, nine patients with FUS-ALS, and three healthy controls. Fujimori used a method called direct neurosphere conversion with chemical pretreatment (CdNS) (Fujimori et al., 2017). The researchers reported that they could detect signs of degeneration in ALS motor neurons differentiated using this protocol sooner than they could using cells grown by other methods.
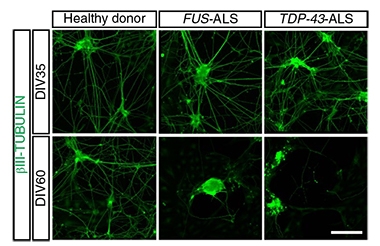
Transition to Degeneration.
After 35 days in culture (top panel), motor neurons had outstretched neurites. By day 60 (bottom panel), neurites from ALS patients had vanished. [Courtesy of Fujimori et al., Nature Medicine, 2018.]
Between 35 and 45 days in culture, the FUS- and TDP-43-ALS neurons leaked lactate dehydrogenase and expressed other markers of apoptosis, and they accumulated stress granules, a common pathology in ALS. TDP-43 and FUS also cropped up in protein inclusions during that time frame. Starting at around 40 days in culture, neurites stopped growing and started shriveling, while neurite outgrowth continued to flourish in motor neurons from healthy donors. Gene expression analysis at day 40 revealed that compared with motor neurons from healthy donors, FUS- and TDP-43 ALS motor neurons shared 689 differentially expressed genes. Some of them belong to pathways previously implicated in ALS while others, unexpectedly, were linked to dopamine synthesis and degradation.
The researchers dubbed this 10-day degenerative period the “transition phase.” They screened a library of more than 1,200 previously approved drugs banked at Keio University for their ability to stave it off. At day 40 of culture, the researchers treated the cells with the drugs, then five days later they measured neurite length, LDH release, numbers of stress granules, and protein inclusions. They found nine drugs that dose-dependently reduced each disease phenotype in the transition phase in both the FUS- and TDP-43-ALS motor neurons. Ropinirole emerged as the top hit in the TDP-43 cases, and the second place hit among FUS cases.
Previously approved to treat Parkinson’s disease and restless leg syndrome, ropinirole activates dopamine D2 (D2R) receptors. To determine if this explained its benefits, the researchers added a D2R antagonist to the neurons as well. This only partially blocked ropinirole’s neuroprotective effects, suggesting both D2R-dependent and -independent mechanisms might play a role. Two structural analogs of ropinirole—PPX and RPPX— scavenge reactive oxygen species (ROS) in mitochondria. RPPX, aka dexpramipexole, was even tested in a Phase 3 clinical trial for ALS, but did not slow the disease (Jan 2013 conference news; Cudkowicz et al., 2013). Fujimori investigated whether ropinirole, too, would scavenge radicals. Indeed, the researchers found that ROS increased in TDP-43- and FUS-ALS motor neurons compared with cells from healthy donors, and that ropinirole treatment prevented this rise in ROS better than did either PPX or RPPX.

Just a Phase? Using four phenotypic markers of disease (labeled on right), researchers zeroed in on a transition phase (green shading) marking the path toward degeneration. Solid dots represent the emergence of each phenotype; triangular boxes represent the severity. [Courtesy of Fujimori et al., Nature Medicine, 2018.]
Would ropinirole’s benefits extend to models of sporadic ALS? To find out, the researchers generated motor neuron cultures from 32 SALS patients. Before adding ropinirole, the researchers first identified the transition phase of each cell line, as they had done with the non-SOD1 FALS cultures. As gauged by the first signs of halted neurite growth and LDH leakage, the researchers found a wide variation in the timing of the transition phase among SALS motor neurons, ranging from about 30–60 days. This window was much broader than for FALS-derived neurons and, notably, came earlier in cells derived from donors with faster disease progression, suggesting these cell culture models captured the clinical severity of disease. Based on how rapidly each model progressed to the phase transition in culture, the researchers classified each motor neuron sample into one of four clusters, ranging from rapid to moderate progression.
The researchers then tested ropinirole on motor neurons from 24 SALS cases. When added at the transition phase, it suppressed neurite retraction, abnormal protein aggregation, and cytotoxicity in most of the cell lines. It blocked apoptosis and ROS elevation in 16 of the 24. The researchers also took stock of the transcriptomes of four representative SALS models around the time of the phase transition, finding changes in genes involved in inflammatory pathways and dopamine regulation in three of the samples that had responded to ropinirole. In the one sample which did not respond, the transcriptome aligned more closely with those from SOD1-ALS motor neurons.
The researchers concluded that ropinirole could make a promising therapeutic candidate for sporadic ALS, as well as non-SOD1 FALS. While it is still unclear how the drug works, they predicted that multiple pathways, including dopamine signaling, mitochondrial function, and cholesterol synthesis, might be involved. They also contended that iPSC-derived motor neuron cultures could be used as a way to predict whether a given patient might respond to a drug.
“Ropinirole is an interesting candidate for drug repurposing, but the authors already note that a similar—albeit less effective—compound failed Phase 3 clinical trials,” Sterneckert wrote. He also pointed out that not every patient’s motor neurons responded to treatment. “Maybe it is possible to use their approach to identify responding patients for clinical testing, which might increase success rates over previous trials, but iPS cell modeling on that kind of scale would still be a considerable challenge.”—Jessica Shugart
References
News Citations
- Hereditary Diseases: A Natural Fit For iPSC Modeling
- Stem Cell Screen Points to ALS Disease Target
- Chicago—ALS Clinical Trials: New Hope After Phase 3 Setbacks
Therapeutics Citations
Paper Citations
- Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, Rouleau GA, Drapeau P. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 2011 Aug;7(8):e1002214. PubMed.
- Da Cruz S, Bui A, Saberi S, Lee SK, Stauffer J, McAlonis-Downes M, Schulte D, Pizzo DP, Parone PA, Cleveland DW, Ravits J. Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol. 2017 Jul;134(1):97-111. Epub 2017 Feb 28 PubMed.
- Fujimori K, Matsumoto T, Kisa F, Hattori N, Okano H, Akamatsu W. Escape from Pluripotency via Inhibition of TGF-β/BMP and Activation of Wnt Signaling Accelerates Differentiation and Aging in hPSC Progeny Cells. Stem Cell Reports. 2017 Nov 14;9(5):1675-1691. Epub 2017 Oct 26 PubMed.
- Cudkowicz ME, van den Berg LH, Shefner JM, Mitsumoto H, Mora JS, Ludolph A, Hardiman O, Bozik ME, Ingersoll EW, Archibald D, Meyers AL, Dong Y, Farwell WR, Kerr DA, EMPOWER investigators. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet Neurol. 2013 Nov;12(11):1059-67. Epub 2013 Sep 23 PubMed.
External Citations
Further Reading
Primary Papers
- Fujimori K, Ishikawa M, Otomo A, Atsuta N, Nakamura R, Akiyama T, Hadano S, Aoki M, Saya H, Sobue G, Okano H. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat Med. 2018 Oct;24(10):1579-1589. Epub 2018 Aug 20 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.