Newest ALS/FTD Gene Keeps Spotlight on Stress Granules
Quick Links
In the August 16 Neuron, researchers led by Rosa Rademakers, Mayo Clinic, Jacksonville, Florida, and Paul Taylor, St. Jude Children’s Research Hospital, Memphis, Tennessee, report that mutations in the gene TIA1 cause amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD). Their study hits a familiar refrain: Like several other ALS genes, TIA1 encodes an RNA-binding protein found in stress granules. Mutations in TIA1 impair stress granule function and promote the accumulation of toxic TDP-43, a hallmark of the diseases, the researchers find.
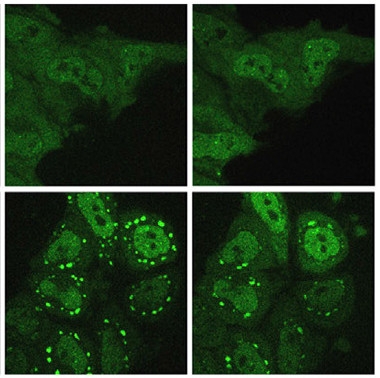
Persistent Granules.
HeLa cells that express A381T TIA1 (bottom) have more stress granules (bright green) at 80 minutes (left) and 120 minutes (right) after heat shock than do control cells (top). [Courtesy of Rosa Rademakers and Neuron.]
TIA1 joins a growing list of RNA-binding proteins that instigate neurodegeneration leading to ALS/FTD, reinforcing the central role of stress granules in that process. Alzforum covered part of the work when Taylor presented it earlier this year at a meeting on liquid-liquid phase transition in Leuven, Belgium (May 2017 conference news).
Rademakers and Taylor collaborated with Ian Mackenzie’s group at the University of British Columbia, Vancouver, Canada. Co-first authors Mackenzie, Alexandra Nicholson, and Mohona Sarkar in Rademakers’ lab first uncovered a potential role for TIA1 in a family with multiple cases of ALS and FTD, and no known disease-causing mutations. Sequencing of all the protein coding genes of two affected members—a woman and her niece—found that they shared rare mutations in 15 genes. A missense variant in TIA1, P362L, stood out. First, TIA1 structurally and functionally resembles other ALS disease genes, such as TDP-43, hnRNPA1, and FUS, and mutations in all these affect protein aggregation in stress granules, where TIA1 localizes. Second, the P362L mutation sat in the prion-like tail of the protein, a region of low amino acid complexity. Low-complexity domains (LCD) play a critical role in protein phase transitions that spark stress granule formation. Last but not least, a TIA1 mutation in the LCD was known to cause a proteinopathy in the form of Welander distal myopathy, a muscle-wasting disease that features TDP-43 aggregates.
Deciding to focus on TIA1, the investigators sequenced the gene in 1,039 other cases of ALS and 3,036 healthy controls. In six patients they found five additional mutations in the TIA1 LCD domain. None of these turned up in healthy controls. The researchers believe the TIA1 mutations are pathogenic and based on these numbers, Rademakers estimates that they could account for one in 50 cases of familial, and as many as one in 200 cases of sporadic ALS, on par with other rare ALS/FTD genes such as VCP and profilin. TIA1 joins a growing list of genes that cause multisystem proteinopathies, striking the nervous system, muscle, or bone.
Carriers of TIA1 mutations shared a special type of pathology. In five cases for which there were autopsy results, the investigators found unusually large, round, and dense TDP-43 inclusions in motor neurons. These “huge” lesions can be found in sporadic ALS, but rarely, said Rademakers. “All of our five patients had them and they had a lot of them,” she told Alzforum. This shared pathology convinced her that the scientists had in fact discovered a new genetic cause of ALS/FTD.
Taylor’s group then analyzed the effects of two of the TIA1 ALS mutations, P362L and A381T, and the E384K distal myopathy mutation, on protein behavior in vitro. All three altered the biophysical properties of the protein, increasing its propensity to undergo liquid-liquid phase separation in vitro. Scientists believe phase separation precedes stress granule formation. While the mutations did not affect stress granule assembly in cells in response to heat shock, after that stress passed the stress granules formed with mutant protein hung around much longer than those that incorporated wild-type TIA1. In addition, TDP-43 recruited to the persistent stress granules rapidly became immobile and insoluble, suggesting a means whereby the mutants promote TDP-43 aggregation and neurodegeneration.
“It’s particularly interesting that disease-causing mutations promoted phase separation. ALS-associated mutations in other genes don’t. They promote fibril formation, which we think is a later step in the process,” said Lindsay Becker, a doctoral student in the lab of Aaron Gitler at Stanford University. “This provides good evidence that phase separation is involved in ALS.”—Pat McCaffrey
References
Alzpedia Citations
News Citations
Further Reading
No Available Further Reading
Primary Papers
- Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, Matchett BJ, Mittag T, Temirov J, Hsiung GR, Krieger C, Murray ME, Kato M, Fryer JD, Petrucelli L, Zinman L, Weintraub S, Mesulam M, Keith J, Zivkovic SA, Hirsch-Reinshagen V, Roos RP, Züchner S, Graff-Radford NR, Petersen RC, Caselli RJ, Wszolek ZK, Finger E, Lippa C, Lacomis D, Stewart H, Dickson DW, Kim HJ, Rogaeva E, Bigio E, Boylan KB, Taylor JP, Rademakers R. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron. 2017 Aug 16;95(4):808-816.e9. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Boston University School of Medicine
The article by Mackenzie et al. adds a new RNA-binding protein to the list of RNA-binding proteins that are linked to ALS and frontotemporal dementia. In one sense the article adds to the growing body of literature showing that dysfunction of RNA-binding proteins contributes to the pathophysiology of ALS. The mutations in TIA1 appear to fit a pattern seen over and over in which mutations in low-complexity domains alter the mobility of TIA1 in the phase-separated mode, and increase the tendency of TIA1 to undergo liquid-liquid phase separation. Previous studies with other proteins suggest that the phase-separated RNA-binding proteins exhibit a tendency to aggregate into insoluble amyloid fibrils. TIA1 also exhibits this tendency, which is increased by the disease causing mutations.
The pathology that occurs in vivo is that associated with TDP-43 inclusions. Previously we demonstrated that TDP-43 co-associates with TIA1 in stress granules in cells and in vivo (Liu-Yesucevitz et al., 2010). The Mackenzie team was able to reproduce the evidence of TDP-43 co-localization in cells. They observed that mutations in TIA1 increase formation of TDP-43 stress granules and aggregated TDP-43. They were unable to reproduce the co-localization of TDP-43 with TIA1 in human tissues; this is unfortunate, but could represent weaknesses of TIA1 commercial antibodies. TIA1 detection is sensitive to fixation time and is hard to detect with fixation longer than 48 hours. However, the group also states that they did not observe evidence of TIA1 aggregation by biochemistry, which would be an observation that would be largely independent of the quality of the antibodies.
In another sense, the article presents a surprising finding because the group presents that the mutations increase the tendency of TIA1 to aggregate, and increase the tendency of the associated pathological protein, TDP-43, to aggregate, but presents no evidence that TIA1 is aggregating in vivo, nor any evidence that TIA1 co-localizes with TDP-43 or any other pathology in vivo. This contrasts with prior work by Hackman and colleagues showing that a different mutation in TIA1 causes a myopathy and formation of TIA1 aggregates (Hackman et al., 2013). This raises two possibilities: 1) TIA1 is inducing TDP-43 aggregation through an indirect mechanism, or 2) the right conditions for modeling and detecting the mechanisms of TIA1 aggregation have yet to be identified.
TIA1 is one of the core nucleating RNA-binding proteins. My laboratory originally started studying TIA1 because we surveyed other core nucleating RNA-binding proteins (e.g., TIA1, TTP, G3BP1 with comparison to RNA-binding proteins such as DCP1, which are found in P-bodies) and observed that TIA1 elicited the strongest response for TDP-43 and later for tau protein (Liu-Yesucevitz et al., 2010; Vanderweyde et al., 2016; Vanderweyde et al., 2012). The current study provides some genetic validation for the hypothesized connection between TDP-43 and TIA1, but also raises many questions. For the moment, the most parsimonious conclusion is that the genetic link of TIA1 to ALS demonstrates its likely role in the biology of ALS, but has yet to identify the mechanism underlying the role.
References:
Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Vanderwyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5(10):e13250. PubMed.
Hackman P, Sarparanta J, Lehtinen S, Vihola A, Evilä A, Jonson PH, Luque H, Kere J, Screen M, Chinnery PF, Åhlberg G, Edström L, Udd B. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol. 2013 Apr;73(4):500-9. Epub 2013 Feb 11 PubMed.
Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PE, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, Ivanov P, Abisambra JF, Steffen M, Li H, Petrucelli L, Wolozin B. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 2016 May 17;15(7):1455-1466. Epub 2016 May 6 PubMed.
Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, Duff K, Wolozin B. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. 2012 Jun 13;32(24):8270-83. PubMed.
Universite de Montreal, CRCHUM
This is a very nice paper with clear experimental evidence that TIA-1 mutant proteins have reduced mobility, and alter stress granule disassembly. The finding that TIA-1 mutations alter stress granule disassembly reaffirms the idea that maintaining correct stress granule dynamics is a key consideration in ALS therapy development. The added finding that the delayed disassembly of stress granules may contribute to the formation of distinct TDP-43 cytoplasmic inclusions is intriguing, and may offer a mechanism for how these inclusions arise. Moreover, it is reasonable to consider a potential feed forward amplification loop, such that as cytoplasmic accumulation proceeds, accompanied by eventual nuclear depletion of TDP-43, there will be a subsequent loss of G3BP1, which further compromises stress granule dynamics.
Stanford University
Mackenzie, Rademakers, and colleagues have uncovered a new ALS-associated gene: TIA1. TIA1 is an RNA-binding protein that is a key component of stress granules, which are liquid-like membraneless organelles that form during cellular stress. A number of other stress granule proteins, such as TDP-43, have also been associated with ALS. TDP-43 forms pathological aggregates in 97 percent of ALS patients. Researchers hypothesize that RNA-binding proteins concentrating within liquid-like stress granules can nucleate insoluble, solid-like TDP-43 aggregates seen in disease. In line with this hypothesis, Mackenzie and colleagues find that ALS-associated mutations in TIA1 impair stress granule dissociation. Additionally, the authors demonstrate TDP-43 within stress granules is quite immobile and insoluble compared to typical stress granule proteins, which are highly motile and easily dissociate.
One of the most interesting findings of the paper is that ALS-associated mutations in TIA1 also promote phase separation of TIA1 in vitro. ALS-associated mutations in other RNA-binding proteins promote the transition from a liquid droplet state to a solid, fibril state. Mutations in TIA1 seem to affect an earlier step in this sequence by facilitating phase separation, in addition to impairing the dynamic properties of the liquid droplets and promoting the formation of fibrils. This difference in mechanism might explain why TIA1 mutation carriers have somewhat unique TDP-43 pathology compared to other ALS patients. This paper provides further evidence that stress granules dynamics are important in the ALS disease cascade and likely will be an effective therapeutic target.
Make a Comment
To make a comment you must login or register.