Death in a Dish: Astrocytes from ALS Patients Flick Necroptosis Switch in Motor Neurons
Quick Links
For astrocytes from patients with sporadic amyotrophic lateral sclerosis (sALS), killing motor neurons seems just like riding a bike—once they know how to do it, they appear to never forget. Researchers led by Serge Przedborski at Columbia University in New York reported in the February 6 Neuron online that even after a month by themselves in a culture dish, such astrocytes rapidly trigger necroptosis, a form of programmed cell death, once they encounter motor neurons. The study could provide a novel way to screen for drug candidates in human cells and yield new treatment strategies for ALS.
“The astrocytes’ ability to maintain this toxic phenotype is quite striking,” said Rick Livesey of the University of Cambridge in England. He was not involved in the work. “This study has opened up a bunch of mechanistic questions that can now be addressed.” The most important of these, Livesey said, are which astrocyte factors hasten necroptosis in motor neurons, and whether this mechanism causes disease in vivo.
ALS manifests in both familial and sporadic forms. About 9 percent of people with the disease have familial ALS (fALS), and mutations in superoxide dismutase 1 (SOD1) occur in 20 percent of these cases (see Jan 2014 news story). Although this equates to only a small fraction of ALS patients overall, many studies have relied upon mouse models that express mutant SOD1. In 2003, researchers led by Don Cleveland at the University of California, San Diego, implicated astrocytes in the death of motor neurons when they showed that limiting expression of the mutant dismutase to these non-neuronal cells still brought on motor neuron disease. In 2007 Przedborski and others confirmed this non-cell autonomy when they extended the observation to mouse cell cultures (see Apr 2007 news story). Other studies have since investigated the same phenomenon in human cells, but have still relied upon SOD1 expression (see Dec 2008 news story). To nail down whether astrocytes from sALS patients can kill motor neurons independently of SOD1, Przedborski decided to start with primary cells. The alternative would be to generate human astrocytes from stem cells, but that could confound the results, he said.
Joint first authors Diane Re and Virginia Le Verche isolated astrocytes from postmortem motor cortex and spinal cord tissue of six sALS patients and 15 controls (see image below). After a month of coddling, astrocytes dominated the cultures. The researchers then mixed these astrocytes with motor neurons derived from human embryonic stem cells. While the neurons thrived when cohabitating with astrocytes from non-sALS controls, their numbers started to plummet after just four days in culture with sALS astrocytes. By 14 days, more than half had perished. Other types of neurons were resistant to the death signals delivered by sALS astrocytes, and fibroblasts from sALS patients harmed no motor neurons either, indicating that the toxic relationship was specific to sALS astrocytes and motor neurons.
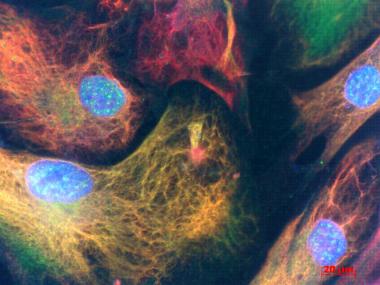
Patients' Astrocytes.
Vimentin (red) and glial fibrillary acidic protein (green) highlight astrocytes cultured from people with sporadic ALS. Cell nuclei are stained blue. [Image courtesy of Diane Re, Columbia University.)
To determine SOD1’s role in this nefarious affair, the researchers knocked down expression of the protein in astrocytes using four different small hairpin RNAs. The treatment failed to protect the motor neurons. Knockdown of astrocyte TDP-43, another protein implicated in familial ALS, did not save them either.
These results contrast a study led by Brian Kaspar at Nationwide Children’s Hospital in Columbus, Ohio, who found that astrocytes derived from neural progenitor cells taken from sALS patients did need SOD1 to kill motor neurons, even though sALS patients harbored no mutations in this gene (see Aug 2011 news story on Haidet-Phillips et al., 2011). Przedborski attributes the discrepancy to multiple technical differences, including the fact that Kaspar’s group used neural progenitor cells to derive astrocytes, not primary cultures. They also did not investigate the mechanism of cell death.
Przedborski and colleagues wanted to know how the motor neurons met their end. Medium from sALS astrocyte cultures triggered death, implicating a soluble factor, but the researchers are still trying to identify it. Because the researchers previously implicated the apoptosis trigger Bax in the demise of motor neurons, they tested whether a Bax inhibitor could protect the cells. It did, but caspase inhibitors that block apoptosis failed, suggesting that this programmed cell death pathway was unlikely to underlie the motor neuron death.
The researchers then searched for a signature of necroptosis, a regulated form of necrosis. Treatment of the cultures with inhibitors of RIP1 or MLKL—two proteins involved in necroptosis—protected the motor neurons. The cells were also spared when the investigators used shRNAs to knock down RIP1 in the motor neurons alone. The same strategies worked to protect mouse motor neurons from mouse astrocytes expressing mutant SOD1, hinting that the same death pathway could be at play in familial and sporadic forms of the disease.
“This work is very interesting and suggests that you could inhibit RIP1 therapeutically to reduce motor neuron loss,” Junying Yuan at Harvard Medical School, Cambridge, Massachusetts, who was not involved in the study, told Alzforum. However, she added that because Bax is not involved in necroptosis, further biochemical analysis of the death pathway in motor neurons is needed. The authors speculate that Bax’s action in the mitochondria of motor neurons somehow switches on necroptosis, rather than apoptosis—a phenomenon documented by other studies (see Artus et al., 2010).
Although the researchers found that SOD1 played no role in astrocyte-mediated killing, they did not rule out the possibility that it does so in cell autonomous death within motor neurons from ALS patients, as non-ALS motor neurons were used in this study. It’s also possible that the dismutase might have a function within astrocytes in vivo that was missed in the short-lived, simplified cell-culture environment, Livesey said.
The Przedborski lab is currently trying to identify the toxic factor the astrocytes release, and to optimize culture conditions to screen for drugs that save motor neurons.—Jessica Shugart
References
News Citations
- Familial ALS More Common Than Thought—Do We Need a New Definition?
- Glia—Absolving Neurons of Motor Neuron Disease
- ALS in a Dish? Studying Motor Neurons from Human Stem Cells
- ALS: Many Disparate Diseases, or Just Two?
Paper Citations
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011 Sep;29(9):824-8. PubMed.
- Artus C, Boujrad H, Bouharrour A, Brunelle MN, Hoos S, Yuste VJ, Lenormand P, Rousselle JC, Namane A, England P, Lorenzo HK, Susin SA. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010 May 5;29(9):1585-99. Epub 2010 Apr 1 PubMed.
Further Reading
Papers
- Meyer K, Ferraiuolo L, Miranda CJ, Likhite S, McElroy S, Renusch S, Ditsworth D, Lagier-Tourenne C, Smith RA, Ravits J, Burghes AH, Shaw PJ, Cleveland DW, Kolb SJ, Kaspar BK. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014 Jan 14;111(2):829-32. Epub 2013 Dec 30 PubMed.
- Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013 Nov;14(11):727-36. Epub 2013 Oct 16 PubMed.
Primary Papers
- Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L, Koolen M, Nagata T, Papadimitriou D, Nagy P, Mitsumoto H, Kariya S, Wichterle H, Henderson CE, Przedborski S. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014 Mar 5;81(5):1001-8. Epub 2014 Feb 6 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.