Could Toning Down Calcineurin Neutralize α-Synuclein?
Quick Links
Excess α-synuclein kills neurons, and part of the reason may well be that the protein causes the cells to overdose on calcium, which hyperactivates the phosphatase calcineurin. In the August 13 Proceedings of the National Academy of Sciences, a paper from the lab of Susan Lindquist, Whitehead Institute for Biomedical Research, Cambridge, Massachusetts, adds evidence that this rampant calcineurin activity mediates α-synuclein toxicity in yeast and neurons. The authors also report that a moderate dose of a calcineurin inhibitor protects the cells, and that there is evidence of calcineurin overactivation in postmortem brains of people who had a synucleinopathy. Lindquist’s group proposes that α-synuclein toxicity could be treated with low doses of the FDA-approved immunosuppressant FK506, which inhibits calcineurin.
“It’s a solid piece of science,” said Giulio Taglialatela, University of Texas Medical Branch at Galveston, who previously implicated calcineurin as a mediator of α-synuclein toxicity (see Martin et al., 2012). FK506 therapy could be relevant to other amyloid-related diseases, he said, as his work and that of others suggests that calcineurin may be activated in Aβ- and prion-related diseases (see Dineley et al., 2007; Reese et al., 2008; Mukherjee et al., 2010; Feb 2010 news story; and Dec 2009 news story).
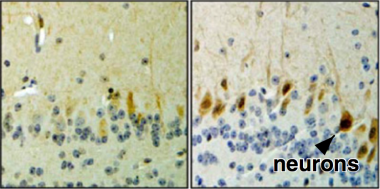
The transcription factor NFATc4 (brown) in cytoplasm in neurons from control mice (left), moves to nuclei (blue) in neurons from transgenic mice overexpressing α-synuclein (right). See text below. Image courtesy of Caraveo et al., PNAS.
Previously, researchers discovered that FK506 protected neurons in models of Parkinson’s disease (PD), though it was not clear why (see Kitamura et al., 1994). The compound binds to FK506 binding protein (FKBP), which then inhibits calcineurin. However, FKBP also functions to refold proteins by converting prolines from the cis to transform, an activity blocked by FK506. Some studies suggested that trans prolines accelerate aggregation of α-synuclein aggregation, (see Gerard et al., 2006), hence researchers focused on inhibiting FKBP as a potential therapeutic (see Guo et al., 2001, and Gerard et al., 2010). However, drugs designed to inhibit FKBP failed to improve Parkinson’s symptoms in humans (see NINDS NET-PD Investigators, 2007). After that, researchers stopped developing them for PD, first author Gabriela Caraveo told Alzforum. With this new study, Lindquist’s group is revisiting the idea that FK506 could treat PD, this time by calming, but not eliminating, calcineurin activity.
When a cell becomes overloaded with α-synuclein, calcium surges into the cytoplasm and binds the protein calmodulin, which then activates calcineurin. The phosphatase plucks phosphates from dozens of different proteins. Lindquist’s group wondered whether dialing back calcineurin activity could reduce the toxicity of α-synuclein.
Caraveo and colleagues used yeast as a model system because calcium pathways are conserved between these cells and humans. In yeast expressing high amounts of α-synuclein, 20- to 30-μg FK506 per milliliter of media protected against synuclein toxicity. However, higher doses (50-μg/ml) killed yeast that overexpressed α-synuclein, while leaving wild-type cells unharmed. Likewise, genetically eliminating calcineurin proved toxic to yeast expressing synuclein, though not to normal cells. Overexpressing calcineurin was also toxic in the synuclein model. These results suggested to the researchers that calcineurin works in Goldilocks fashion—neither too much nor too little, but just the right amount of activity combats α-synuclein toxicity.
Moderate doses of FK506 also protected rat primary cortical neurons and mouse dopaminergic neurons that expressed α-synuclein. In roundworms, partial knockdown of calcineurin with RNAi rescued α-synuclein toxicity.
The authors next went in search of affected downstream pathways of calcineurin. One of the phosphatase’s important substrates is nuclear factor of activated T cells (NFAT), which moves to the nucleus to activate gene expression when it is dephosphorylated. NFAT functions in numerous cell types, including neurons. The researchers wondered if NFAT was activated by excess α-synuclein, and whether FK506 could prevent that. Caraveo looked at the stress-response protein Crz1 in yeast, the functional analog of the mammalian NFAT. Like NFAT, Crz1 is a transcription factor and its activity is suppressed by phosphorylation. Caraveo found more dephosphorylated Crz1 in yeast overexpressing α-synuclein, but adding FK506 prevented that dephosphorylation. That hinted that α-synuclein activates the transcription factor and FK506 keeps it in check, wrote the authors.
In mammals, α-synuclein appeared to affect NFAT pathways in a similar fashion. Transgenic mouse models overexpressing α-synuclein in the brain activated the NFATc4 isoform in neurons and NFATc3 in glia. In postmortem brains from eight patients diagnosed with PD or dementia with Lewy bodies (DLB), NFATc4 showed up more often in neuronal nuclei from the substantia nigra pars compacta, hippocampus, and frontal cortex (see image above). In five control brains, NFATc4 staining remained principally in the cytoplasm.
The results support the idea that calcineurin mediates α-synuclein toxicity, and makes the phosphatase a drug target, wrote the authors. They propose that FK506 be repurposed as a treatment for synucleinopathies. Called Tacrolimus, FK506 has been in clinical use for many years as an immunosuppressant, for example to avoid transplant rejection or to treat eczema. The drug can have serious side effects including infection, vasoconstriction and kidney toxicity. However, Caraveo believes that neuroprotective doses are much lower. Taglialatela agreed that this drug could be developed for neurodegenerative diseases. His group is looking for evidence that FK506 decreases the incidence of AD and PD in patients who have been taking the drugs for a long period.
Chris Norris, University of Kentucky, Lexington, concurred that low-dose FK506 warrants study as a therapy for neurodegeneration. Norris found that NFAT activation correlated with cognitive decline in patients with Alzheimer’s or mild cognitive impairment (see Oct 2009 news story). Other studies have tied NFAT activation to AD (see Hudry et al., 2012). Norris suggested that researchers test whether FK506 preserves motor function and protects dopaminergic neurons in rodent models of α-synucleinopathies. Calcineurin is expressed widely in the body, however, making safe dosing difficult. FK506’s therapeutic index—that is, the dose range at which a drug is both safe and effective—is generally considered narrow. Future therapies might steer FK506 or similar compounds to specific cells to avoid global inhibition, Norris said.
In a related report, another group recently implicated a calcineurin- and NFAT-dependent pathway in α-synuclein toxicity, and reported that inhibiting calcineurin with another inhibitor, cyclosporin A, lessened degeneration of dopaminergic neurons (see Luo et al., 2014).
As a step toward clinical trials in Parkinson’s patients, Caraveo and colleagues are currently wrapping up a study of moderate doses of FK506 in rat models of PD.—Gwyneth Dickey Zakaib
References
Alzpedia Citations
News Citations
- Calcium Hypothesis—Studies Beef Up NFAT, CaN, Astrocyte Connections
- Chicago: NFATs, Calcineurin—Mediators of AD, PD Pathogenesis?
- The Skinny on NFATs—Mediators of Aβ Toxicity?
Paper Citations
- Martin ZS, Neugebauer V, Dineley KT, Kayed R, Zhang W, Reese LC, Taglialatela G. α-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: relevance to human synucleopathic diseases. J Neurochem. 2012 Feb;120(3):440-52. PubMed.
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007 Sep;88(2):217-24. PubMed.
- Reese LC, Zhang W, Dineley KT, Kayed R, Taglialatela G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell. 2008 Dec;7(6):824-35. PubMed.
- Mukherjee A, Morales-Scheihing D, Gonzalez-Romero D, Green K, Taglialatela G, Soto C. Calcineurin inhibition at the clinical phase of prion disease reduces neurodegeneration, improves behavioral alterations and increases animal survival. PLoS Pathog. 2010 Oct 7;6(10):e1001138. PubMed.
- Kitamura Y, Itano Y, Kubo T, Nomura Y. Suppressive effect of FK-506, a novel immunosuppressant, against MPTP-induced dopamine depletion in the striatum of young C57BL/6 mice. J Neuroimmunol. 1994 Mar;50(2):221-4. PubMed.
- Gerard M, Debyser Z, Desender L, Kahle PJ, Baert J, Baekelandt V, Engelborghs Y. The aggregation of alpha-synuclein is stimulated by FK506 binding proteins as shown by fluorescence correlation spectroscopy. FASEB J. 2006 Mar;20(3):524-6. Epub 2006 Jan 12 PubMed.
- Guo X, Dillman JF 3rd, Dawson VL, Dawson TM. Neuroimmunophilins: novel neuroprotective and neuroregenerative targets. Ann Neurol. 2001 Jul;50(1):6-16. PubMed.
- Gerard M, Deleersnijder A, Daniëls V, Schreurs S, Munck S, Reumers V, Pottel H, Engelborghs Y, Van den Haute C, Taymans JM, Debyser Z, Baekelandt V. Inhibition of FK506 binding proteins reduces alpha-synuclein aggregation and Parkinson's disease-like pathology. J Neurosci. 2010 Feb 17;30(7):2454-63. PubMed.
- Hudry E, Wu HY, Arbel-Ornath M, Hashimoto T, Matsouaka R, Fan Z, Spires-Jones TL, Betensky RA, Bacskai BJ, Hyman BT. Inhibition of the NFAT pathway alleviates amyloid β neurotoxicity in a mouse model of Alzheimer's disease. J Neurosci. 2012 Feb 29;32(9):3176-92. PubMed.
- Luo J, Sun L, Lin X, Liu G, Yu J, Parisiadou L, Xie C, Ding J, Cai H. A calcineurin- and NFAT-dependent pathway is involved in α-synuclein-induced degeneration of midbrain dopaminergic neurons. Hum Mol Genet. 2014 Dec 15;23(24):6567-74. Epub 2014 Jul 22 PubMed.
External Citations
Further Reading
Papers
- Cavallucci V, Berretta N, Nobili A, Nisticò R, Mercuri NB, D'Amelio M. Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer's disease. Neuromolecular Med. 2013 Sep;15(3):541-8. PubMed.
- Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol. 2009 Feb;4(2):481-508. PubMed.
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007 Sep;88(2):217-24. PubMed.
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007 Feb 1;53(3):337-51. PubMed.
- Rozkalne A, Hyman BT, Spires-Jones TL. Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol Dis. 2011 Mar;41(3):650-4. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.