Brain Imaging Suggests Aβ Unleashes the Deadly Side of Tau
Quick Links
Aβ nudges tau out of the hippocampus and into the cortex, where the previously benign microtubule-associated protein turns into a neuronal killer. At least, this is the hypothesis that gains support from the use of brain imaging to estimate tau accumulation and neurodegeneration in people with or without Aβ deposits in the brain. Published July 25 in JAMA Neurology, the findings are the latest of several recent reports using the tau tracer [18F]-AV-1451. While some of those linked tau deposition to abnormal cerebrospinal fluid (CSF) tau and Aβ, dips in brain glucose metabolism, and AD progression, the new analysis is the first published to correlate AV-1451 uptake with loss of brain volume. The researchers, led by Beau Ances at Washington University in St. Louis, report that in people who have Aβ accumulation in the brain, tau deposition correlates tightly with brain atrophy. Even among some cognitively normal people harboring Aβ deposits, tau accumulated in the cortex, casting cortical tau as a potentially useful disease staging tool.
Until recently, anything scientists knew about tau filaments in the human brain came from postmortem analyses. Those suggested that neurofibrillary tangles arose in the entorhinal cortex, then spread into the hippocampus and the rest of the cerebral cortex as AD progressed (see Braak and Braak, 1991; Delacourte et al., 1999). PET tracers have since allowed researchers to monitor the distribution of tau aggregates in living people. Thus far, cross-sectional analyses indicate that tau filaments build up in the brain in much the same order as reported from neuropathology studies, only appearing in the cortex if Aβ plaques are present (see Mar 2016 news). Tangles also seem to accumulate in metabolically sluggish areas of the brain, and in functionally impaired regions; for example, AD patients who struggle finding words tend to amass more tau in the left hemisphere of the cortex, which processes language (see Ossenkoppele et al., 2016). Other reports have linked cortical tau deposition to cognitive decline and AD progression, and also to characteristic changes in CSF biomarkers (see Johnson et al., 2016; May 2016 news; and Gordon et al., 2016).
“What was still missing after all of this research is how tau deposition directly relates to neurodegeneration,” Ances told Alzforum.
First author Liang Wang and colleagues sought to fill in that gap by investigating the relationship between tau deposition, Aβ, and brain atrophy. First, they wanted to confirm that tau imaging can track disease progression. The researchers conducted AV-1451 PET scans of 59 participants, 42 of whom had undergone lumbar puncture and been designated as Aβ-positive or -negative based on the concentration of Aβ42 in their CSF. Of these 42 patients, 35 were cognitively normal, including 14 people positive for Aβ. The remaining seven were diagnosed with AD, although one of them tested negative for Aβ. People with AD had higher levels of tau deposition in the hippocampus and in several “AD cortical signature regions.” Originally identified as areas of the cortex where thinning correlated with disease progression, atrophy in these signature regions also correlated with tau accumulation in both autopsy and CSF biomarker studies (see Dickerson et al., 2009; Vemuri et al., 2008; and Wang et al., 2015).
Using a cutoff of 1.19 in the standardized uptake value ratio (SUVR) for AV-1451 in the AD cortical signature regions, the researchers could distinguish healthy controls who had normal CSF Aβ from people with AD. A fraction of cognitively normal people who tested positive for Aβ had SUVRs above the cutoff, suggesting they might be on the verge of conversion to AD, Ances said. This indicated that tau imaging could be used as a staging tool to determine who among cognitively normal people will soon develop AD, he said.
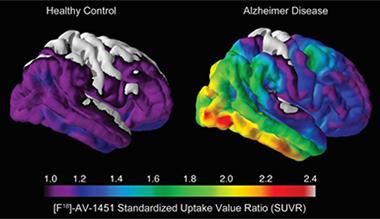
Tau Takeover.
Compared with healthy controls (left), people with AD (right) harbored large amounts of insoluble tau in the medial temporal lobe and neocortical areas. [Image courtesy of Beau Ances, Washington University.]
After confirming that the tracer seemed to track with disease progression, the researchers next teased apart the relationships between Aβ plaques, tau deposition, and brain volume. They found that unlike in the AD cortical signature regions—where tau was higher in Aβ-positive people than in Aβ-negatives—in the hippocampus, tau deposition did not vary with Aβ status (though it trended higher in people with Aβ).
What about brain volume? Compared with cognitively normal people, those with AD had smaller hippocampi and thinning in cortical regions associated with the disease. Tau levels in the hippocampus and cortex inversely correlated with hippocampal volume and cortical thickness, respectively. In the hippocampus, the relationship only held in people who had abnormal CSF Aβ. However, in the cortex, the tau deposition coupled tightly with atrophy regardless of Aβ status. The strength of this relationship between tau and neurodegeneration maxed out in the medial temporal lobe, and waned moving outward into the inferior and lateral temporal, and ultimately parietal areas. This pattern tracked closely with the hypothetical path of neurofibrillary tangle propagation proposed previously based on neuropathology (see Braak and Braak, 1991). Together, the findings suggested to the researchers that in the absence of Aβ, hippocampal tau seems insufficient for neurodegeneration. They propose that the accumulation of Aβ somehow transforms this benign hippocampal tau into a toxic entity, which then proceeds to kill neurons in the hippocampus and in the cortical regions to which it then spreads, an idea that has gained momentum (see Aug 2015 conference news; Jan 2016 conference news).
Christopher Rowe of the University of Melbourne in Australia found this suggestion intriguing, but said that the study may have been too small to determine whether neurodegeneration depended upon the conversion of tau into a more toxic form, or simply on there being more of the protein present. That hippocampal neurodegeneration among people with similar levels of hippocampal tau only occurs when Aβ enters the picture supports the idea of conversion to some toxic form of tau. However, Rowe pointed out that the level of tau in the hippocampus of Aβ-positive people did trend higher than that in Aβ-negative people, suggesting tau concentration may still be germane. “A larger study is needed to clarify this potentially very important issue,” he wrote.
Mark Mintun of Avid Radiopharmaceuticals in Philadelphia agreed. “Maybe Aβ does transform tau into a malignant form, but we can’t conclude that with this data alone,” he said. However, he pointed out that the correlations between tau and neurodegeneration were impressive, and called the study icing on the cake of previous studies linking cortical tau to the transition to clinical AD.
“The findings support the idea that this tau tracer will be a powerful way to monitor neurodegeneration during clinical trials,” Mintun added. He is currently heading a clinical trial aimed at validating [18F]-AV-1451 as a bona fide imaging agent for tau, in which researchers will compare tau imaging in patients close to death to postmortem neuropathology afterwards (see clinicaltrials.gov).
“Mapping brain tau in relation to measures of neurodegeneration is the beginning of more precisely staging the disease,” wrote William Jagust of the University of California, Berkeley, in an editorial that accompanied the paper. “Preclinical AD may ultimately be staged by relating tau to these neurodegenerative biomarkers, and the neocortical localization of tau may be a harbinger of incipient neurodegeneration and cognitive symptoms. This staging may be crucial in selecting people for participation in clinical trials of drugs that are directed at either Aβ or tau,” he added.—Jessica Shugart
References
News Citations
- Tau PET Aligns Spread of Pathology with Alzheimer’s Staging
- On Multiple Marker Analysis, Tangles Track Best With Functional Decline
- New Imaging Data Tells Story of Travelling Tau
- Tau Takes Center Stage at 10th Human Amyloid Imaging Conference
Paper Citations
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-59. PubMed.
- Delacourte A, David JP, Sergeant N, Buée L, Wattez A, Vermersch P, Ghozali F, Fallet-Bianco C, Pasquier F, Lebert F, Petit H, Di Menza C. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease. Neurology. 1999 Apr 12;52(6):1158-65. PubMed.
- Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016 May;139(Pt 5):1551-67. Epub 2016 Mar 8 PubMed.
- Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, Mormino E, Chhatwal J, Amariglio R, Papp K, Marshall G, Albers M, Mauro S, Pepin L, Alverio J, Judge K, Philiossaint M, Shoup T, Yokell D, Dickerson B, Gomez-Isla T, Hyman B, Vasdev N, Sperling R. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016 Jan;79(1):110-9. Epub 2015 Dec 15 PubMed.
- Gordon BA, Friedrichsen K, Brier M, Blazey T, Su Y, Christensen J, Aldea P, McConathy J, Holtzman DM, Cairns NJ, Morris JC, Fagan AM, Ances BM, Benzinger TL. The relationship between cerebrospinal fluid markers of Alzheimer pathology and positron emission tomography tau imaging. Brain. 2016 Aug;139(Pt 8):2249-60. Epub 2016 Jun 10 PubMed.
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, Grodstein F, Wright CI, Blacker D, Rosas HD, Sperling RA, Atri A, Growdon JH, Hyman BT, Morris JC, Fischl B, Buckner RL. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009 Mar;19(3):497-510. PubMed.
- Vemuri P, Whitwell JL, Kantarci K, Josephs KA, Parisi JE, Shiung MS, Knopman DS, Boeve BF, Petersen RC, Dickson DW, Jack CR. Antemortem MRI based STructural Abnormality iNDex (STAND)-scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage. 2008 Aug 15;42(2):559-67. PubMed.
- Wang L, Benzinger TL, Hassenstab J, Blazey T, Owen C, Liu J, Fagan AM, Morris JC, Ances BM. Spatially distinct atrophy is linked to β-amyloid and tau in preclinical Alzheimer disease. Neurology. 2015 Mar 24;84(12):1254-60. Epub 2015 Feb 25 PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, McConathy J, Cairns NJ, Fagan AM, Morris JC, Ances BM. Evaluation of Tau Imaging in Staging Alzheimer Disease and Revealing Interactions Between β-Amyloid and Tauopathy. JAMA Neurol. 2016 Sep 1;73(9):1070-7. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Austin Hospital
The authors report that tau imaging with F-18 AV1451 can accurately distinguish healthy controls from AD. While important as part of validation of a new technique, this in itself is not very useful when an interview can do the job just as well in these straightforward, clinically defined cases. More important is the suggestion by the authors that amyloid may modify hippocampal tau to a more toxic form and that this then drives neurodegeneration of that structure. While an intriguing suggestion, the data is not very convincing due to small sample size. The suggestion relies on the finding that there is no significant difference in hippocampal tau in those with amyloid compared to those without, but that hippocampal volume loss is much greater when amyloid is present. But looking at the data points in figure 3, there is an indication that there is more tau in those with amyloid, suggesting the study may be underpowered and failed to detect this. So, it may be that amyloid is associated with more hippocampal tau and that the neurodegeneration is tau dose-related rather than due to conversion of tau to a more toxic form by amyloid. A larger study is needed to clarify this potentially very important issue.
UCLA Neuropsychiatric Institute
A speculative, but intriguing, implication of these findings is that Aβ can facilitate the membrane channel forming properties of tau. Our recent paper on tau showed that it was capable of forming non-selective and potentially destructive ion channels in lipid membranes, but it was less active as a channel-former than Aβ. It is possible that Aβ may interact with tau to catalyze its entry into membranes, leading to cellular energy depletion, dysfunction, and eventually cell death. While we have some hints that this interaction takes place in vitro, demonstrating it in vivo could be very informative and perhaps lead to agents that block this deadly amyloid interaction.
References:
Patel N, Ramachandran S, Azimov R, Kagan BL, Lal R. Ion Channel Formation by Tau Protein: Implications for Alzheimer's Disease and Tauopathies. Biochemistry. 2015 Dec 22;54(50):7320-5. Epub 2015 Dec 7 PubMed.
Make a Comment
To make a comment you must login or register.