PET Tracers Enlighten at Neurology Conference
Quick Links
Imaging technologies have transformed the study of neurodegenerative diseases by giving scientists a glimpse at what is happening inside the brains of living patients. At the 66th annual meeting of the American Academy of Neurology, held 26 April to 3 May in Philadelphia, researchers touted the potential of several positron emission tomography (PET) tracers to improve diagnoses. A tau tracer under development looks promising for characterizing several types of frontotemporal lobar degeneration (FTLD), while amyloid imaging distinguishes effectively between FTLD and Alzheimer’s disease in young dementia patients, speakers said. Meanwhile, fluorodeoxyglucose (FDG) PET was confirmed once again to reveal dampened brain metabolism in people who carry one or more ApoE4 alleles, the main genetic risk factor for sporadic AD, though surprisingly, this effect occurred in people young and old, with or without amyloid deposits.
“We are in a revolutionary era of neurodegenerative disease research,” Brad Dickerson Massachusetts General Hospital, Boston, told Alzforum. “[New imaging technologies] will completely change the way we approach these diseases.”
These days the biggest buzz surrounds the potential of tau tracers, since tau pathology marks numerous neurodegenerative diseases. Several companies are developing such tracers (see Jan 2013 conference story; Aug 2013 conference story; Sep 2013 news story). At AAN, Dickerson reported preliminary results from imaging with Eli Lilly and Company’s T807 in about 15 patients with various tauopathies. These included behavioral-variant FTLD, frontotemporal aphasias, and the movement disorder progressive supranuclear palsy (PSP).
In all cases, the brain regions that T807 lit up most strongly matched the regions with the greatest atrophy and hypometabolism in that particular disease, Dickerson said. That means these three major brain imaging modalities used in living patients are aligned. In behavioral-variant FTLD patients, the frontal, insular, and anterior temporal cortices gave the strongest T807 signal, whereas Broca’s aphasia patients bound the most tracer in the inferior frontal and middle temporal gyri. These patterns correspond to the areas of greatest degeneration in each disease. Moreover, they are distinct from the profile seen in Alzheimer’s, where T807 lights up temporal regions. The findings suggest that T807 binding reflects tau deposits, but final confirmation will have to come from postmortem studies, Dickerson said.
How early in disease might a tau signal appear? Dickerson described one cognitively healthy participant who carried the tau mutation P301L, which causes frontotemporal dementia. Although the man was at least 10 years away from his expected age of symptom onset, his T807 signal was mildly elevated in the same regions of the brain that light up in symptomatic patients. The results hint that tau tracers could help identify people at preclinical disease stages.
One concern emerged from the data. Only about 40 percent of clinically diagnosed FTLD cases are tauopathies; many others are characterized instead by TDP-43 deposits. Ideally, a tau tracer would allow scientists to distinguish between these proteins. However, in this study, a participant who carried a progranulin mutation, which causes TDP-43 pathology, had an elevated T807 signal. “That was a major surprise,” Dickerson told Alzforum. This raises the question of whether T807 might react non-specifically with TDP-43; alternatively, this patient might have concurrent tau pathology. In ongoing work, Dickerson is scanning more people with different predicted pathologies to try to nail down how specific the tracer is. Eventually, he will validate in-vivo findings against autopsy results.
Despite the questionable specificity, other researchers found these results exciting. “In patients with PSP and tau mutations, the data looks very promising,” said Gil Rabinovici at the University of California, San Francisco.
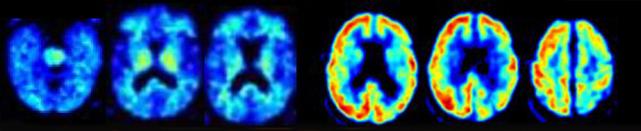
A patient clinically diagnosed with Alzheimer’s was negative for amyloid imaging (left scans), but positive by FDG PET (right scans). Postmortem pathology identified the disease as corticobasal degeneration. [Image courtesy of Gil Rabinovici.]
FTLD cases can be difficult to distinguish from early onset AD. Amyloid imaging may help clinicians make this distinction, Rabinovici said in his AAN talk. Currently, many clinicians use FDG PET for differential diagnosis, because insurers such as the Centers for Medicare and Medicaid Services (CMS) reimburse for this scan. By contrast, CMS does not yet cover clinical use of amyloid imaging (see Jul 2013 news story). Many researchers believe amyloid imaging would do a better job than FDG PET of distinguishing these diseases. Rather than compare patterns of glucose use, as FDG does, amyloid tracers detect a specific, defining pathology present in AD that is typically absent in FTD. Researchers are currently gathering evidence to try to persuade the CMS to change its stance (see Oct 2013 news story).
For his part, Rabinovici compares the diagnoses made after FDG and PIB amyloid scans in patients being treated at memory clinics. Previously, he reported that amyloid imaging was more reliable between different raters than FDG PET, as well as more sensitive and equally specific, in a cohort of about 100 patients (see Rabinovici et al., 2011). In response, CMS noted that this data was promising, but lacked autopsy confirmation. At AAN, Rabinovici reported autopsy results from the first 44 patients in this cohort. Twenty-eight turned out to have had FTLD, 12 AD, 3 mixed FTLD and AD, and one had had prion disease.
Amyloid imaging correctly identified all the amyloid and mixed-pathology patients as having AD, and ruled out the prion patient. In the FTLD group, however, four patients had neuritic plaques that showed up on PIB PET scans, resulting in an AD diagnosis. Pathologists judged that these plaques were unlikely to have contributed to dementia, and called the cases FTLD. This gave amyloid imaging a sensitivity of 100 percent and a specificity of about 90 percent for distinguishing AD from FTLD. By contrast, FDG reads misclassified several AD and FTLD cases, and had an overall sensitivity of about 86 percent and specificity of 79 percent. All numbers favored amyloid imaging, but the cohort was too small to show statistical significance. Nonetheless, Rabinovici said the data indicate that amyloid imaging is at least as good as FDG PET for distinguishing FTLD and AD.
“The data definitely support the reimbursement of amyloid imaging in the FTLD versus AD scenario,” Rabinovici told Alzforum. Misdiagnosis can be harmful, because AD medications such as cholinesterase inhibitors and memantine have been found to worsen outcomes for FTLD patients in clinical trials, Rabinovici noted. However, he stressed that amyloid scans should be interpreted by experts, and only after a comprehensive clinical evaluation has been conducted. “Scans can be misinterpreted. A positive amyloid scan doesn’t rule out FTD, for example. I don’t think this technique is ready for widespread use in primary care,” he said.
While FDG PET may become passé for differential diagnosis of FTD versus AD, this technique has many other applications. At AAN, David Knopman at the Mayo Clinic in Rochester, Minnesota, reported using it to study metabolic changes in more than 800 cognitively normal adults in the Mayo Clinic Study of Aging. His data were published March 11 in Neurobiology of Aging. He found a modest decline with age in most cortical and sub-cortical brain regions. Previous studies had conflicted on this point, with some showing no age-related changes. The current study is much larger than most prior studies and used more rigorous methods, making the results more definitive, Knopman claimed.
Knopman also wanted to know how an ApoE4 allele would affect metabolism. The 209 study participants who carried at least one ApoE4 allele had lower glucose uptake than non-carriers did in the posterior cingulate, precuneus, lateral parietal, and inferior temporal regions. Intriguingly, these regions typically atrophy in AD. Knopman’s data confirm previous FDG PET findings in ApoE4 homozygotes by Eric Reiman and colleagues at Banner Alzheimer’s Institute, Phoenix, and others (see, e.g., Reiman et al., 1996; Reiman et al., 2004; Lehmann et al., 2014). The new data also extends earlier FDG PET observations on ApoE. In particular, it shows that age has no bearing. The ApoE4 effect looms as large in 30-something adults as in people in their 70s. Knopman said this surprised him. The magnitude of the drop in metabolism seen in carriers is about one-fourth of that seen in AD patients versus age-matched controls.
Notably, glucose metabolism in ApoE4 carriers was lower regardless of whether people had amyloid in their brain, agreeing with some previous data (see Dec 2012 news story). This suggests that ApoE4 may contribute to AD through some other mechanism, Knopman said. In future work, Knopman will compare FDG scans with both amyloid and tau imaging at multiple age ranges to try to gain more insight into the pathological mechanisms.
Other researchers found the results intriguing. Rabinovici wrote to Alzforum, “While ApoE4 is clearly a risk factor for amyloid aggregation, there is increasing evidence that ApoE4 also increases the risk for AD via a multitude of Aβ-independent pathways, some of which traverse the entire lifespan (see excellent review by Huang and Mucke, 2012, and interesting model proposed by Jagust and Mormino, 2011). This has major implications as the field develops strategies for AD treatment and prevention in E4 positive individuals.” (See full comment below.)—Madolyn Bowman Rogers
References
News Citations
- HAI—Spotlight on Tau Tracers at Human Amyloid Imaging Meeting
- Tau Tracers Shine at Boston Conference
- Tau Tracer May Light Up All Tauopathies
- Coverage Denial For Amyloid Scans Riles Alzheimer’s Community
- Alzheimer’s Community Mobilizes to Show Benefits of Amyloid Scans
Paper Citations
- Rabinovici GD, Rosen HJ, Alkalay A, Kornak J, Furst AJ, Agarwal N, Mormino EC, O'Neil JP, Janabi M, Karydas A, Growdon ME, Jang JY, Huang EJ, Dearmond SJ, Trojanowski JQ, Grinberg LT, Gorno-Tempini ML, Seeley WW, Miller BL, Jagust WJ. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology. 2011 Dec 6;77(23):2034-42. PubMed.
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996 Mar 21;334(12):752-8. PubMed.
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004 Jan 6;101(1):284-9. PubMed.
- Lehmann M, Ghosh PM, Madison C, Karydas A, Coppola G, O'Neil JP, Huang Y, Miller BL, Jagust WJ, Rabinovici GD. Greater medial temporal hypometabolism and lower cortical amyloid burden in ApoE4-positive AD patients. J Neurol Neurosurg Psychiatry. 2013 Aug 21; PubMed.
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012 Mar 16;148(6):1204-22. PubMed.
- Jagust WJ, Mormino EC. Lifespan brain activity, β-amyloid, and Alzheimer's disease. Trends Cogn Sci. 2011 Nov;15(11):520-6. PubMed.
Other Citations
Further Reading
Primary Papers
- Knopman DS, Jack CR Jr, Wiste HJ, Lundt ES, Weigand SD, Vemuri P, Lowe VJ, Kantarci K, Gunter JL, Senjem ML, Mielke MM, Roberts RO, Boeve BF, Petersen RC. 18F-fluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol Aging. 2014 Sep;35(9):2096-106. Epub 2014 Mar 11 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UCSF
This study by Knopman and colleagues from Mayo Clinic demonstrates that decreases in brain glucose metabolism are independently associated with both advancing age and ApoE4 genotype in cognitively normal individuals. The effects of age appeared quite global, while the ApoE4 effect was relatively specific to AD-vulnerable regions. Strengths of the study include the large sample size, and the use of partial volume correction to demonstrate that metabolic decline is independent of atrophy. A relative weakness is the small number of ApoE4 homozygotes—this is expected in studies of normal aging, but precluded investigation of a dose effect for ApoE4.
A striking finding in the study is that the effect of ApoE4 was present across a broad age range (30-95), and was independent of amyloid burden as assessed by PiB-PET. While ApoE4 is clearly a risk factor for amyloid aggregation, there is increasing evidence that ApoE4 also increases the risk for AD via a multitude of Aβ-independent pathways, some of which traverse the entire life-span (see review by Huang and Mucke, 2012, and interesting model proposed by Jagust and Mormino, 2011). This has major implications as the field develops strategies for AD treatment and prevention in E4-positive individuals.
References:
Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012 Mar 16;148(6):1204-22. PubMed.
Jagust WJ, Mormino EC. Lifespan brain activity, β-amyloid, and Alzheimer's disease. Trends Cogn Sci. 2011 Nov;15(11):520-6. PubMed.
View all comments by Gil RabinoviciMayo Clinic
That [18F]-T807 uptake in FTLD syndromes co-localizes with structural changes seen on MRI scans demonstrates the sensitivity of this PET ligand for the pathology of FTLD. This finding has implications for early diagnosis as well as for tracking disease progression and potential treatment responses at the initial stages of FTLD pathology. Ideally, [18F] T807 uptake would differentiate between FTLD syndromes with TDP versus those associated with tau deposition. However, the specificity of [18F] T807 binding to the tau protein in FTLD needs to be further investigated with in-vitro and in-vivo studies and with autopsy confirmation.
National Institute on Aging
The interesting paper by Knopman et al. referred to in this report convincingly demonstrates statistically significant age reductions in brain glucose metabolism as measured with PET FDG in a large cohort (806) of cognitively normal subjects ranging in age from 30 to nearly 100. The conclusions differ somewhat from findings by Ibanez et al. in a study that examined statistically insignificant age changes in resting-state, atrophy-corrected glucose metabolism comparing 13 older men (55 to 82 years old) with 11 younger men (22 to 34 years).
While the difference certainly can be ascribed to the Knopman paper's greater power due to its larger numbers and many older subjects, other methodological differences deserve to be noted. One is that the Ibanez paper determined absolute glucose metabolic rates by quantifying the arterial input function to calculate metabolism, where the Knopman study did a ratio analysis (regional to global incorporated radioactivity) to estimate glucose metabolism. Additionally, the Ibanez study excluded hypertensive subjects, whereas the Knopman paper does not mention subject blood pressure. The exclusion is relevant because Salerno et al. reported that even well-treated hypertensive subjects had reduced brain glucose metabolism compared with controls, and the prevalence of hypertension increases with aging.
Both the Knopman and Ibanez papers agree that cerebral atrophy occurs with aging, and another study using PET FDG reported reduced correlations between regional metabolic rates, suggesting functional disconnection as well.
References:
Knopman DS, Jack CR Jr, Wiste HJ, Lundt ES, Weigand SD, Vemuri P, Lowe VJ, Kantarci K, Gunter JL, Senjem ML, Mielke MM, Roberts RO, Boeve BF, Petersen RC. 18F-fluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol Aging. 2014 Sep;35(9):2096-106. Epub 2014 Mar 11 PubMed.
Ibáñez V, Pietrini P, Furey ML, Alexander GE, Millet P, Bokde AL, Teichberg D, Schapiro MB, Horwitz B, Rapoport SI. Resting state brain glucose metabolism is not reduced in normotensive healthy men during aging, after correction for brain atrophy. Brain Res Bull. 2004 Mar 15;63(2):147-54. PubMed.
Salerno JA, Grady C, Mentis M, Gonzalez-Aviles A, Wagner E, Schapiro MB, Rapoport SI. Brain metabolic function in older men with chronic essential hypertension. J Gerontol A Biol Sci Med Sci. 1995 May;50(3):M147-54. PubMed.
Horwitz B, Duara R, Rapoport SI. Age differences in intercorrelations between regional cerebral metabolic rates for glucose. Ann Neurol. 1986 Jan;19(1):60-7. PubMed.
The Medical and Pharmacological Research Center Foundation
I would like to comment on this interesting article that refers to our previous studies (Yanase et al., 2005; Samuraki et al., 2012) on similar topics.
Knopman et al. observed a modest but significant age-related FDG decline in many brain regions, including areas that are known to be affected in AD, such as the posterior cingulate/precuneus, even after correction for atrophy. In our prior study, also using atrophy-corrected FDG PET (Yanase et al., 2005), the area of age-related FDG decline was confined to fewer regions, including the anterior cingulate and inferior frontal gyrus. There are at least two differences between the studies that may have affected the results. First, their study included a much larger number of cognitively normal individuals (n=806) than our study (n=139), which is certainly beneficial for unmasking significant regions that could not be detected in smaller studies. Second, the atrophy-correction algorithms were different. Where our study used a three-compartment method that accounted for the effect of partial-volume averaging between gray and white matter in addition to the diluting effects of CSF spaces, Knopman and colleagues used a two-compartment method. Because each method has its advantages and disadvantages, it would be worthwhile to see whether the results are consistent over different methodologies.
In their study, there was an APOE ε4-associated FDG decline in AD-vulnerable regions, which was independent of amyloid burden as measured by PiB PET. In our other prior study (Samuraki et al., 2012), however, the prevalence of AD-like imaging abnormalities, such as an FDG decline in the posterior cingulate/precuneus or hippocampal atrophy, did not differ among APOE ε4 carriers with normal cognitive function and non-carriers. In that study, we did not use atrophy correction for FDG PET, because we expected that uncorrected FDG signal would reflect both metabolism and atrophy due to partial-volume effect, and therefore may serve as a more sensitive marker of AD-associated changes. This should particularly be the case in individuals with posterior cingulate/precuneus atrophy, which is known to occur in a subset of AD patients (Shima et al., 2012). That they found the opposite—FDG decline in AD-vulnerable regions was more significant with atrophy correction than without—is interesting. I assume that the use of atrophy correction may have lessened atrophy-related variability in measured FDG signal and thereby increased the statistical power to detect APOE ε4-associated alterations, although this hypothesis needs to be addressed in further studies.
In addition to the atrophy-correction issue, the magnitude of APOE ε4-associated FDG decline that they found seems to be much smaller than that typically seen in AD patients.
Further, our study participants were much younger (mean age: 53.6 years for the ε4 carriers and 53.5 years for the non-carriers) than those in the Knopman et al. study (median age: 76 years); the majority of our participants were 30 to 60 years old: there were no significant differences between ε4 carriers and non-carriers in that age group in their study. The APOE ε 4-associated FDG decline may be more difficult to detect in such young subjects. All these circumstances, alone or in combination, may have contributed to the somewhat discordant observations between the studies.
From a pathophysiological perspective, it would be interesting to see how APOE ε4-associated FDG decline interacts with structural alterations measured by MRI. In our study, a cognitively normal individual with AD-like FDG decline did not necessarily have hippocampal atrophy, and vice versa. In this regard, PET and structural MRI combination would be useful for more comprehensive understanding of APOE ε4-related metabolism-atrophy interaction.
Finally and most importantly, it should be noted that many factors other than APOE ε4, such as diabetes mellitus, are likely to be involved in the development of AD-like imaging signatures in cognitively normal individuals, which has nicely been demonstrated in a recent study by the same research group (Roberts et al., 2014). In our study a subset of ε4 non-carriers showed AD-like imaging signatures, further supporting this notion. Non-ApoE factors can be genetic or non-genetic, and need to be identified in further studies.
In summary, this study provides an important clue for better understanding of interaction between normal aging and AD-related changes in the brain. At the same time, questions still remain to be addressed.
References:
Knopman DS, Jack CR Jr, Wiste HJ, Lundt ES, Weigand SD, Vemuri P, Lowe VJ, Kantarci K, Gunter JL, Senjem ML, Mielke MM, Roberts RO, Boeve BF, Petersen RC. 18F-fluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol Aging. 2014 Sep;35(9):2096-106. Epub 2014 Mar 11 PubMed.
Roberts RO, Knopman DS, Cha RH, Mielke MM, Pankratz VS, Boeve BF, Kantarci K, Geda YE, Jack CR Jr, Petersen RC, Lowe VJ. Diabetes and elevated hemoglobin A1c levels are associated with brain hypometabolism but not amyloid accumulation. J Nucl Med. 2014 May;55(5):759-64. Epub 2014 Mar 20 PubMed.
Samuraki M, Matsunari I, Chen WP, Shima K, Yanase D, Takeda N, Matsuda H, Yamada M. Glucose metabolism and gray-matter concentration in apolipoprotein E ε4 positive normal subjects. Neurobiol Aging. 2011 Dec 19; PubMed.
Shima K, Matsunari I, Samuraki M, Chen WP, Yanase D, Noguchi-Shinohara M, Takeda N, Ono K, Yoshita M, Miyazaki Y, Matsuda H, Yamada M. Posterior cingulate atrophy and metabolic decline in early stage Alzheimer's disease. Neurobiol Aging. 2011 Aug 18; PubMed.
Yanase D, Matsunari I, Yajima K, Chen W, Fujikawa A, Nishimura S, Matsuda H, Yamada M. Brain FDG PET study of normal aging in Japanese: effect of atrophy correction. Eur J Nucl Med Mol Imaging. 2005 Jul;32(7):794-805. Epub 2005 Mar 10 PubMed.
View all comments by Ichiro MatsunariUniversity of Colorado School of Medicine
I agree with the comments from Dr. Rabinovici regarding ApoE possibly playing a multifactorial role in AD risk over the lifespan. In addition to the excellent articles by Huang and Mucke, and Jagust and Mormino, I would like to mention two reviews that I was involved with that provide expanded discussion of the potential link of apoE to energy metabolism (Wolf et al., 2013; Wolf et al., 2013). Additionally, I believe it is fundamental that cellular/molecular studies be utilized to determine potential mechanisms underlying the findings in these brain imaging studies.
References:
Wolf AB, Valla J, Bu G, Kim J, LaDu MJ, Reiman EM, Caselli RJ. Apolipoprotein E as a β-amyloid-independent factor in alzheimer's disease. Alzheimers Res Ther. 2013 Sep 3;5(5):38. PubMed.
Wolf AB, Caselli RJ, Reiman EM, Valla J. APOE and neuroenergetics: an emerging paradigm in Alzheimer's disease. Neurobiol Aging. 2013 Apr;34(4):1007-17. PubMed.
Make a Comment
To make a comment you must login or register.