DIAN: What Sayeth the Regulator? Q&A With Rusty Katz
Quick Links
When the Dominantly Inherited Alzheimer Network (DIAN) convened its Pharma Consortium on 14 April 2012 in Washington, D.C, its scientists intended to brief the Food and Drug Administration on how far DIAN’s work has advanced to date (see Part 1). Their other goal was to obtain feedback from the FDA on the study designs DIAN trialists have proposed. To this end, DIAN Pharma Consortium (PC) members asked the advice of Russell Katz, who directs the agency’s neurology products division. A paraphrased and abbreviated Q&A of the conversation follows.
DIAN PC: Are there regulatory concerns in our choice of a so-called seamless design, where a biomarker phase runs into a longer Phase 3 trial, which then uses cognitive outcome measures?
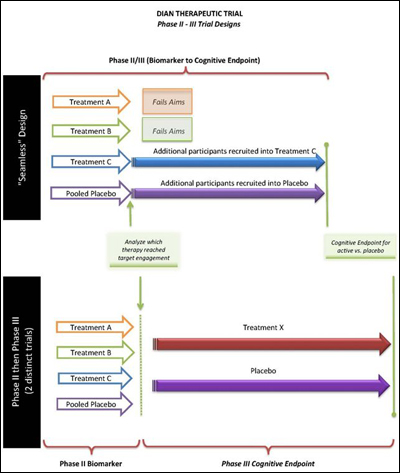
Seamless trial design proposed by DIAN Therapeutic Trials Unit. View larger image. Image credit: DIAN Therapeutic Trials Unit
Russell Katz: I see no particular regulatory challenges to a seamless design. You need to address blinding questions and assure us that a type 1 error is not inflated in the envisioned design. It’s best to lay that out prospectively. That has been done before. One more point: Let’s assume arm A gets dropped and arm C continues; presumably, then, participants in arm A will be moved to C. That is not a problem.
DIAN PC: Is the intra-individual rate of change an acceptable endpoint if the parent DIAN study data are used as the run-in baseline data for the biomarker and cognitive endpoint trials?
Russell Katz: Conceptually, the agency has no objections to using intra-individual change. How exactly you are planning to analyze it will be important.
DIAN PC: For example, what if a person’s 20 percent per year decline in DIAN decreased to a 0 percent per year decline during the trial?
Russell Katz: You’d have to show it is statistically significant, but that is an impressive change.
DIAN PC: Can DIAN use pre-trial data on biomarkers, CSF, imaging, clinical, and cognitive to determine a change, or individual rate of change? For example, can we use data that have been collected in DIAN as a baseline for evidence that a drug has changed PIB retention?

Hypothetical change in brain amyloid before and during treatment trial. View larger image. Image credit: DIAN Therapeutic Trials Unit
Russell Katz: Yes, this would be fine, assuming data collection was done post-randomization as it was done pre-randomization, i.e., during DIAN.
DIAN PC: Would the FDA concur, as indicated in November 2010 (see ARF related news story), that for individuals with autosomal-dominant AD and pre-manifest disease, a subtle clinical change in conjunction with an effect on a surrogate or even multiple surrogate markers could be acceptable for approval?
Russell Katz: Yes, we are open to that, for example, an effect on a composite cognitive measure.
DIAN PC: Would one such study be considered sufficient evidence?
Russell Katz: We are open to that, too. But the effect has to be robust, on the primary outcome, on the secondary outcomes. It has to be unassailable.
DIAN PC: If two drugs show benefit in the biomarker trial, are there any problems from a regulatory viewpoint of taking both forward (i.e., same placebo group, etc.)?
Russell Katz: I see no significant regulatory problem there.
DIAN PC: Would change in CDR-SOB as a single primary efficacy measure be suitable for registration?
Russell Katz: We are open to that. We have not signed off on that formally, but have told sponsors to flesh this out for us. We recognize that very early on, when people are asymptomatic, the usual rules are difficult to apply.
DIAN PC: Would a time-to-change outcome measure or a single cognitive measure or composite be sufficient for registration?
Russell Katz: Again, that is possible.
DIAN PC: How might these different endpoints affect the labeled indication if the trial is positive?
Russell Katz: It’s hard to know exactly what the label would say. We would be reluctant to grant a disease-modifying claim just on the basis of a clinical outcome. If you have a change on a biomarker and on a clinical outcome, then we contemplate granting a disease-modifying claim. What kind of a claim a positive trial would support also depends on whether all people who carry different mutations respond in the same direction.
DIAN PC: Based on FDA feedback to date, the agency prefers one IND holder, i.e., DIAN, with multiple product INDs, rather than the alternative of having multiple IND holders using the same protocol. What are the obstacles to having multiple IND holders?
Russell Katz: It is hard to imagine that we would accept multiple IND holders for the same trial. It would be a complicated trial and an administrative nightmare to deal with multiple sponsors. The regulations say that the sponsor is responsible for the conduct of the trial. There has to be one entity. With I-SPY, for example, that is the Foundation of the National Institute of Health (FNIH). I cannot imagine more than one IND holder.
DIAN PC: Under a single IND scenario, DIAN will need to meet certain obligations. DIAN will be responsible for IND amendments, safety reports, etc. Could these obligations, particularly those related to the reporting of serious adverse events (SAEs) to the FDA, be delegated or transferred to DIAN’s pharma partners?
Russell Katz: No. You can’t ask folks who are not the sponsor to handle an obligation of this sort that you are proposing. Amendments, safety, etc., have to come to us through the sponsor. Suppose there is a safety issue. It has to come to us; it has to be disseminated to all. How is that possible with multiple IND holders? I have asked about this idea internally, but we think it is not workable. I know it is of concern to some pharma partners. We are happy to sit down with the group to try to figure this out, but we are fairly clear the answer is no.
Russell Katz: What is the objection to a single IND holder?
DIAN PC member, speaking for the member's respective company, not the entire committee: It is not scientific or medical. Some companies are concerned about risk to their drug if they are not controlling the interaction with the FDA, especially if it is a drug that is under FDA review for approval based on other trials the company is also doing. For example, DIAN has not done much SAE reporting in a registration clinical trial. When an SAE comes in, sometimes it is difficult to determine if it is related to treatment, especially in such an unusual cohort.
Russell Katz: I don’t understand why it can’t happen under one IND holder. I have not heard anything today that would require multiple INDs, and see problems on our side coordinating separate studies with separate INDs.—Gabrielle Strobel.
This is Part 2 of a series. See also Part 1. Read a PDF of the series.
References
News Citations
- DIAN Grows, Gets Ready for Therapeutic Trials
- London: What Regulators Say About Trials in Familial AD
Other Citations
{kind=link}
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.