Excess α-Synuclein Sends Synapses Sputtering
Quick Links
Though excess amounts of α-synuclein have long been known to cause familial Parkinson disease and to characterize sporadic PD, what this protein actually does in individual neurons has remained a mystery. In a cell biology study published June 16 in the Journal of Neuroscience, researchers led by Subhojit Roy of the University of California, San Diego, propose that several-fold increases of α-synuclein can trigger a slew of pathological changes leading to synaptic dysfunction. The findings appear, at one level, consistent with a January Neuron paper in which Robert Edwards, University of California, San Francisco, and colleagues report waning synaptic transmission with modestly elevated α-synuclein (Nemani et al., 2010). Both studies suggest that a little extra synuclein causes big problems with transmitter release. However, the UCSD group describes this as one of many detriments of excess synuclein, whereas the UCSF team identified a highly specific defect in reclustering of synaptic vesicles with little else going wrong in their synuclein-overexpressing neurons. While these incompatibilities may stem from the studies’ different model systems and experimental approaches, the authors and others agree that both reports provide valuable clues—at a single-cell level in more physiologically relevant settings—about α-synuclein’s effects on the synaptic machinery.
In people, tripling the amount of wild-type α-synuclein protein causes severe PD symptoms and pathology (Singleton et al., 2003 and ARF related news story), and mere doubling produces serious clinical and pathological changes (Ikeuchi et al., 2008; Fuchs et al., 2008; Farrer et al., 2004). Yet in the lab, scientists have been largely stuck with PD models that overexpress α-synuclein at whopping non-physiological levels.
The new studies try to bridge the gap by looking at what happens to neurons with modestly elevated (two- to threefold) α-synuclein. In the JNS paper, first author David Scott and colleagues made transgenic mice expressing low levels of human wild-type synuclein tagged with green fluorescent protein. These mice developed spatial memory problems by six months of age, and had about 2.5 times more α-synuclein than did wild-type littermates. To study the effects of excess α-synuclein on individual cells, the researchers cultured hippocampal neurons from transgenic mice “and followed the green glow over time,” Roy told ARF. “We were completely agnostic. We looked at them each week to see what was different [from hippocampal neurons of wild-type controls].”
The researchers saw that GFP-synuclein clustered at presynaptic terminals (see image below) without causing overt synapse loss. After three weeks in culture, though, transgenic neurons showed evidence of pathological change, as they formed proteinase K-resistant aggregates and racked up phosphorylated serine-129. In electrophysiological experiments, α-synuclein-overexpressing neurons struggled to release neurotransmitter, and ultrastructural analyses revealed strange features within the underperforming cells. “We saw weird-looking, large vesicular structures five to 10 times larger than what you’d normally see in a synapse,” Roy said. Furthermore, some presynaptic terminals—which the researchers dubbed “vacant synapses”—appeared to lack four key synaptic proteins: vesicular-SNARE protein (VAMP-2), Piccolo, synapsin, and amphiphysin. Attempting to relate these in-vitro observations with human disease, the researchers immunostained autopsy brain tissue from dementia with Lewy body (DLB) patients and found that many α-synuclein-positive synapses also lacked synapsin.
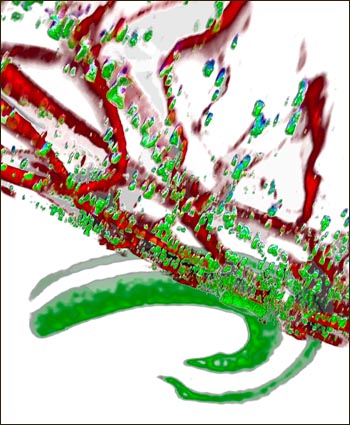
Synaptic α-synuclein
α-synuclein (green) clusters at synaptic terminals and co-localizes with the somatodendritic marker MAP-2 (red) in hippocampal neurons of transgenic mice with 2.5-fold overexpression of wild-type α-synuclein. Image credit: David Scott
In the Neuron paper, first author Venu Nemani and colleagues also found reduced transmitter release resulting from mild overexpression of α-synuclein, but did not see most of the other α-synuclein-induced changes described by the UCSD group. Instead, by directly imaging the synaptic vesicle cycle in primary hippocampal neurons transfected with wild-type human α-synuclein to two- to threefold normal protein levels, the UCSF researchers reported a highly specific defect—recycling vesicles were failing to cluster near synaptic release sites. With help from UCSF colleague Roger Nicoll, the scientists found similarly reduced transmitter release in hippocampal slices of transgenic mice with threefold α-synuclein overexpression. Furthermore, culture experiments showed that the effect on transmitter release requires the N-terminal membrane-binding domain of α-synuclein.
Considering both papers, “the real issue is what α-synuclein is actually doing,” Edwards told ARF. “When you overexpress α-synuclein, does that produce a gain of its normal function? What [Roy] is arguing is that it produces a gain of its abnormal function. These are fundamentally different views.” This question should find resolution through ongoing analysis of knockout mice lacking all three α-synuclein isoforms. “If these animals show an increase in transmitter release, it would strongly suggest that the effects we observe involve a gain in the normal function of α-synuclein,” Edwards noted (see full comment below).
Roy finds the data of Edwards’s team “direct and convincing” and noted “it is possible that a “reclustering” defect is a major pathology induced by excess α-synuclein with additional smaller defects in other aspects of the synaptic machinery.” However, in light of the JNS data and prior α-synuclein studies in yeast and mice, along with well-known pleiotropic effects of other proteins implicated in neurodegeneration (e.g., tau, amyloid), “we favor the view that α-synuclein has diverse effects on the synaptic release apparatus,” Roy said.
Kostas Vekrellis, Biomedical Research Foundation of the Academy of Athens, Greece, thinks the different conclusions may derive from time and conformation of α-synuclein in the experimental systems used by the two recent studies. Roy’s team found pathological changes in cultured neurons with sustained α-synuclein expression (i.e., transgenic mice), whereas Edwards and colleagues examined neurons with shorter α-synuclein expression (i.e., transfection) and which did not seem to be aggregated, Vekrellis noted in an e-mail to ARF (see full comment below).
One issue the current studies leave unresolved is how impaired transmitter release might lead to Lewy body pathology or neurodegeneration. However, given that some of the most vulnerable neurons in PD are loaded with synaptic terminals and thus highly active, “it is not difficult to imagine that compromising the ability of these neurons to release transmitter might force them to work even harder than demanded by their normal role in brain, and that, after 50 or 60 years, they are left with axonal pathology (Galvin et al., 1999) and LBs,” wrote James Surmeier, Feinberg School of Medicine at Northwestern University, Chicago, Illinois, in a commentary accompanying the study by Edwards and colleagues (Surmeier, 2010).—Esther Landhuis
References
News Citations
Paper Citations
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010 Jan 14;65(1):66-79. PubMed.
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003 Oct 31;302(5646):841. PubMed.
- Ikeuchi T, Kakita A, Shiga A, Kasuga K, Kaneko H, Tan CF, Idezuka J, Wakabayashi K, Onodera O, Iwatsubo T, Nishizawa M, Takahashi H, Ishikawa A. Patients homozygous and heterozygous for SNCA duplication in a family with parkinsonism and dementia. Arch Neurol. 2008 Apr;65(4):514-9. PubMed.
- Fuchs J, Tichopad A, Golub Y, Munz M, Schweitzer KJ, Wolf B, Berg D, Mueller JC, Gasser T. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J. 2008 May;22(5):1327-34. PubMed.
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004 Feb;55(2):174-9. PubMed.
- Galvin JE, Uryu K, Lee VM, Trojanowski JQ. Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci U S A. 1999 Nov 9;96(23):13450-5. PubMed.
- Surmeier DJ. alpha-Synuclein at the synaptic gate. Neuron. 2010 Jan 14;65(1):3-4. PubMed.
Further Reading
Papers
- Surmeier DJ. alpha-Synuclein at the synaptic gate. Neuron. 2010 Jan 14;65(1):3-4. PubMed.
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003 Oct 31;302(5646):841. PubMed.
Primary Papers
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010 Jun 16;30(24):8083-95. PubMed.
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010 Jan 14;65(1):66-79. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of California, Irvine
This elegant study by Scott et al. takes advantage of primary neuron cultures from α-synuclein-GFP-transgenic mice to examine the effects of modest α-synuclein overexpression on presynaptic proteins. They find convincing evidence that α-synuclein can diminish levels of several critical presynaptic proteins involved in exocytosis and endocytosis. The authors also detect significant reductions in miniEPSC frequency, diminished presynaptic exocytosis, and altered vesicle size by EM in α-synuclein-overexpressing neurons. Thus, physiologically relevant increases in α-synuclein produce robust functional consequences that closely mimic those observed in animal models of endocytic protein deficiency.
The authors point out that similar effects on presynaptic proteins have recently been shown following Aβ oligomer exposure (Parodi et al., 2010), suggesting a possible common mechanism of synaptic dysfunction between AD and synucleinopathies. It is intriguing to speculate that this potential shared mechanism of synaptic dysfunction may play a role in the acceleration of cognitive decline and aggressive disease course in patients and transgenic mice that co-exhibit both AD and Lewy body pathologies (Olchney et al., 1998; Clinton et al., 2010).
References:
Parodi J, Sepúlveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG. Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol Chem. 2010 Jan 22;285(4):2506-14. PubMed.
Olichney JM, Galasko D, Salmon DP, Hofstetter CR, Hansen LA, Katzman R, Thal LJ. Cognitive decline is faster in Lewy body variant than in Alzheimer's disease. Neurology. 1998 Aug;51(2):351-7. PubMed.
Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, Laferla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010 May 26;30(21):7281-9. PubMed.
View all comments by Mathew Blurton-JonesUniversity of California, San Diego
Our goal in this study was to try connecting the dots between two key pathologic events: modestly elevated α-synuclein levels within the neuron and the ultimate synaptic dysfunction. We used a cell-biological approach that allowed us to analyze and quantify thousands of α-synuclein overexpressing boutons. Based on the data, we suggest a cascade of pathologic events initiated by modest elevations of α-synuclein and culminating in synaptic damage. Studies by Nemani et al. focus on the effects of elevated α-synuclein on specific steps within the synaptic release/recycling machinery by directly imaging the synaptic cycle in α-synuclein transfected neurons.
First, it is important to emphasize that using a variety of methods, both studies show at a single-neuron level that the overall synaptic defect induced by modestly elevated α-synuclein is an inhibition of neurotransmitter release. Thus, collectively, these studies provide a firm pathologic role that can be attributed to α-synuclein overexpression. The studies by Nemani et al. also show a dose-dependent effect of excessive α-synuclein, and strongly implicate the N-terminus of α-synuclein in the pathogenesis. However, while Nemani et al. posit an exclusive impairment of “reclustering” of synaptic vesicles as the solitary defect induced by excessive α-synuclein upon the presynaptic apparatus, our studies suggest that that the effects of elevated α-synuclein on the synaptic apparatus are diverse, including defects in pathways involved in both endo- and exocytosis.
The experiments by Nemani et al. are direct and convincing, and it is possible that a “reclustering” defect is a major pathology induced by excessive α-synuclein, with other minor defects on other aspects of the synaptic machinery as well. As relative newcomers to the field, we have no favorite hypothesis on how α-synuclein causes inhibition of neurotransmitter release. However, given our data and other α-synuclein studies in yeast and mice, and the well-known pleiotropic effects of other well-studied proteins implicated in neurodegeneration (tau, amyloid), we favor the view that α-synuclein has diverse effects on the synaptic release apparatus.
�
The background for our work is that α-synuclein normally localizes to the axon terminal of essentially all neurons, but its role, if any, in neurotransmitter release has remained very unclear. In general, knockout mice have shown either no effect or conflicting effects on synaptic transmission. Increased expression of α-synuclein causes Parkinson disease (PD)—duplication and triplication of the wild-type gene cause severe familial PD, and the protein accumulates in all sporadic PD. In light of this, we wondered what overexpression might do to synaptic transmission. This seemed particularly interesting because overexpression of wild-type α-synuclein in mice actually fails to produce degeneration, and an effect on transmitter release would be easier to interpret in the absence of toxicity.
To understand how α-synuclein affects neurotransmitter release, we used a combination of primary neuronal culture and genetic manipulation in mice. The reason is that, although culture is very powerful to dissect molecular mechanism, it suffers from greater variability and has more potential for artifact than analysis in vivo. By optical imaging, we found that α-synuclein specifically inhibits synaptic vesicle exocytosis, in particular, the extent of exocytosis rather than the rate, with no effect on synaptic vesicle endocytosis. We used a variety of experimental approaches (FM dyes, pHluorin reporter) to document a selective reduction in the size of the synaptic vesicle recycling pool, with no change in the total number of synaptic vesicles. Since previous work has suggested differences between dopamine and other neurons, we also used the primary culture to compare midbrain dopamine neurons with hippocampal glutamate neurons, and found that α-synuclein inhibits transmitter release in both populations.
In collaboration with our lab neighbor Roger Nicoll, we found that hippocampal slices prepared acutely from transgenic mice also show a reduction in transmitter release, establishing the physiological relevance of our findings in culture. Further, we used the culture system to show that the effect on release requires the N-terminal membrane-binding domain of α-synuclein.
The paper by Roy and colleagues also shows that α-synuclein inhibits transmitter release. The effect on synaptic vesicle exocytosis appears different from what we observed, and they also find a variety of other changes that we did not. We observed no effect of α-synuclein on the rate of synaptic vesicle exocytosis; the Roy paper uses FM dyes to suggest an effect on the rate but does not corroborate this with other methods. Surprisingly, Roy et al. use transgenic mice to produce the cultures that serve as the basis for their experiments, but perform little analysis using the mice themselves to corroborate their in-vitro findings in vivo. Rather, they rely on the cultures to pursue biochemical and ultrastructural analysis, finding 1) a major reduction in all synaptic vesicle proteins, including some synaptic boutons without synaptic vesicle proteins that they term “vacant boutons,” although it is a bit difficult to call anything a bouton if there are no synaptic vesicles in it; and 2) gross changes in presynaptic ultrastructure by EM.
It is true that culture can be very helpful to elucidate changes at the level of individual boutons. Yet our transgenic mice express α-synuclein in essentially all neurons, so we should also have seen a reduction in synaptic vesicle proteins if there were any. Furthermore, we did observe a reduction in the synapsins, but not other synaptic vesicle proteins, demonstrating that we could detect a biochemical effect of the transgene. We also looked at the transgenic mice by electron microscopy and found a much more specific effect of α-synuclein overexpression—a dispersion of synaptic vesicles away from the release site.
Overall, the Roy paper attributes the effect of α-synuclein on synaptic transmission to “upstream events,” but also states that the effects are pleiotropic and hence suggestive of toxicity. In contrast, we found very selective effects on the transmitter release mechanism and little evidence of toxicity. So the debate comes down to a basic question: Does α-synuclein overexpression produce changes through a gain in the normal function of the protein (as we suggest) or through the gain of an abnormal function (as suggested by Roy et al.)? This is a profound question with direct relevance for the pathogenesis of PD. It is always difficult to tell whether the toxicity observed is relevant to the disease, or just some other kind of injury. That is why we prefer to study a system with selective rather than general effects.
In the end, the question of normal function will be settled through the analysis of α-synuclein knockout mice. At this point, only double-knockout mice have been reported, but triple-knockouts lacking all the isoforms exist and are now being analyzed. If these animals show an increase in transmitter release, it would strongly suggest that the effects we observe involve a gain in the normal function of α-synuclein. Indeed, recent work using double-knockout mice has suggested an increase in dopamine release (Senior et al. 2008). Speaking in more practical terms, it will be difficult to elucidate the toxicity observed by Roy et al. since it is not clear where to begin. In contrast, we think that the selectivity of the defect we observe provides a clear starting point for future investigation.
In summary, both our and Roy's studies observe an effect of α-synuclein on synaptic vesicle exocytosis, but the details and the interpretation are quite different. The extent of our analysis in multiple experimental systems with a range of complementary methods suggests that the effect of α-synuclein is highly specific, and perhaps related to its normal role. However, this conclusion awaits further analysis of α-synuclein knockout mice, and identification of the mechanism by which α-synuclein acts to inhibit synaptic vesicle exocytosis. The identification of a specific effect also provides an entry point for future study of its role in disease as well as normal physiology.
References:
Senior SL, Ninkina N, Deacon R, Bannerman D, Buchman VL, Cragg SJ, Wade-Martins R. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur J Neurosci. 2008 Feb;27(4):947-57. PubMed.
University of Toronto
The work by Scott and colleagues is of great interest as it is trying to pinpoint the molecular details in the synaptic pathology caused by a modest transgenic overexpression of α-synuclein. The authors found that PK-resistant and abnormally phosphorylated α-synuclein tends to accumulate in dysfunctional boutons. They also elegantly demonstrated that such boutons display a gradual reduction in levels of certain endogenous presynaptic proteins. In an attempt to extend their findings to human disease, they looked for and confirmed similar alterations on a DLB brain section.
I think another transgenic model that moderately overexpresses another neuronal protein (e.g., APP) should have been looked at in parallel (to exclude that the effects seen are merely an effect of protein overproduction). Also, more human cases should have been included to verify that the observed differences are truly relevant to disease. Even so, the findings are intriguing, and the described model would be very useful to test effects of heat-shock proteins and other putative rescuing molecules. Moreover, with the emergence of novel antibodies and other detection tools, it will be interesting to relate cellular effects and the distribution of monomeric, oligomeric, and fibrillar α-synuclein accumulating presynaptically and in other parts of the affected neuron.
Effects on neurotransmission by α-synuclein were the subject of study in the paper by Nemani et al. Also here, a modest increase in the intraneuronal levels of α-synuclein was sufficient to cause aberration. More specifically, the N-terminal domain of α-synuclein seems to be critical in mediating an impairment in transmitter release. However, contrary to the study by Roy, this paper did not find any corresponding changes in vesicle numbers or presence of presynaptic proteins (although the experimental set-up was different and the two groups did not study the same proteins).
Taken together, the two studies demonstrate new and interesting aspects of synaptic pathology induced by deposition of pathological α-synuclein. Applying the insights from the study by Nemani et al. on the model presented by Scott et al., it would be of interest to see if the N-terminal part of α-synuclein is responsible for all the aspects described.
UNIVERSITÄTSMEDIZIN GÖTTINGEN GEORG-AUGUST-UNIVERSITÄT
Both papers present evidence that the pathophysiological mechanism in synucleinopathies is not neuronal cell death but a synaptic dysfunction; that is very interesting. With regard to the clinical symptoms in PD, (also PDD and DLB), the synaptic pathology is due to a decrease in neurotransmitter release. The two publications provide us with a link between α-synuclein overexpression and an impairment of vesicle turnover. With this approach, it might be possible to explain the clinical symptoms of PD.
Both papers show that α-synuclein-related pathology is not restricted to dopaminergic neurons.
The conclusion to be drawn from the results of these papers is that PD and DLB research should move away from models of α-synuclein-related toxicity or cell death that can be achieved only by unphysiologically high overexpression of α-synuclein. Rather, research should concentrate on synaptic failure associated with moderately altered α-synuclein levels. The link to α-synuclein aggregation was only drawn in the Scott et al. paper.
�
α-synuclein and Synaptic Failure in PD
α-synuclein has been biochemically and genetically linked to sporadic and familial PD. Mutations or multiplications of the α-synuclein gene cause familial forms of PD (Polymeropoulos et al., 1997; Krüger et al., 1998; Singleton et al., 2003; Zarranz et al., 2004). The aberrant function of α-synuclein is not understood, although there is evidence that abnormal folding and aggregation may play a role and that the toxic α-synuclein species may be oligomeric intermediates. It has been shown that α-synuclein is highly enriched in presynaptic terminals. At this site, it could be acting as a modifier of synaptic vesicle recycling, dopamine storage, and release at nerve terminals. Recent work has also suggested a role for α-synuclein in SNARE-mediated exocytosis at the synapse. In this respect, the synaptic role of α-synuclein is based primarily on the results of knockout studies (Chandra et al., 2005; Larsen et al., 2006). Still, the effects of α-synuclein overexpression on synaptic function had not received much attention. The recent papers by Nemani et al., 2010, and Scott et al., 2010, clearly show that overexpression of full-length α-synuclein at mild levels leads to defects in the synapse, including altered exocytosis, suggesting that this is an early event in the pathogenesis of PD.
In the Nemani et al. study, the authors use a battery of elegant optical and physiological in vitro and in vivo experiments to show, for the first time, that a relatively short time of overexpression of α-synuclein leads to the fast appearance of presynaptic defects in glutamatergic hippocampal neurons and mesencephalic dopaminergic neurons.
What is exciting about this manuscript is that the reduction of neurotransmitter release occurs under only modest overexpression of α-synuclein and in the absence of any aggregation and toxicity. What is more, the effects appear to be dose-dependent. This suggests that the observed effects might reflect early pathologic events in the disease pathway. Detailed kinetic examination further demonstrated that the inhibition did not involve defects in vesicle fusion or a reduction in the number of transmitter-containing vesicles, but rather a specific defect in the synaptic vesicle recycling pathway by preventing the reclustering of vesicles after endocytosis. Interestingly, the effect was specific to membrane-associated mutations of α-synuclein and therefore to the N-terminus of the molecule.
Although the molecular pathway by which overexpression of α-synuclein inhibits synaptic vesicle reclustering was not deciphered in the study of Nemani et al., it is possible that synapsin downregulation observed in α-synuclein overexpressing cells may play a role. Still, a direct physical (inter)action of the proteins was not identified.
To address the question of α-synuclein expression and synapse physiology, and to understand the mechanisms by which α-synuclein expression affects synapse function, Scott et al. set out a series of rigorous quantitative cell biological experiments using a transgenic mouse model in which human α-synuclein was tagged at the C-terminus to GFP (Rockenstein et al., 2005). Unlike the study by Nemani et al., the model of α-synuclein overexpression used by Scott et al. exhibited post-translationally modified and pathologically altered α-synuclein despite the modest levels of expression. Using electrophysiological assessment, the authors elegantly demonstrated a significant reduction in neurotransmitter release and a failure of the presynaptic exocytotic machinery in the synaptic boutons of transgenic mice neurons. Importantly, styryl dye uptake experiments further suggested that the recycling machinery in the transgenic cultures was inoperable, since a significant number of boutons overexpressing α-synuclein had failed to endocytose the dye. Electron microscopy examination of such synapses further revealed variation in the size of synaptic vesicles, including vesicles enlarged in size. Since this phenotype resembled previously reported animal models that lacked presynaptic proteins (Schoch et al., 2001; Nonet et al., 1998; Deitcher et al., 1998), the authors went on to quantitatively evaluate the levels of a number of endocytic and exocytic proteins (including synapsin) in transgenic synapses and found that they were either absent or diminished.
The authors concluded (and rightly so), that multiple exocytic and endocytic pathways are involved in α-synuclein pathogenesis. Significantly, such changes were also observed in human autopsy samples of synucleinopathies (DLB). In this respect, it would have been interesting to see whether and how the distribution of such proteins changes in the vulnerable brain areas during aging. What is important in this study of Scott et al. is the sustained expression of α-synuclein in the model used, and the observation that the protein is pathologically altered. It is very likely that misfolded and/or aggregated α-synuclein have different effects on the synaptic function and morphology. One might also conclude that a long-lasting expression of α-synuclein, as in the case of multiplications of the gene, has different effects on the physiology and morphology of the synapse, to a transient/shorter one as the two studies suggest. Again, the identification of species generated in the two scenarios is in need of further examination.
Another question that arises is whether “aggregated” α-synuclein species have a direct interaction with presynaptic proteins. What would the effect of “oligomeric” modifiers be on the morphology of the vesicles and function of the synapse? Collectively, the findings presented in these studies suggest that PD may result from a “dying back” process. This would be an early event initiated at the site of synapse, stemming from the accumulation and misfolding of α-synuclein and leading to degeneration of basal ganglia axons followed by more widespread denervation. However, it will take many more studies to pinpoint which of the cellular actions of overexpressed α-synuclein lie at the center of synaptic physiology. A critical question that remains open is how a reduction in neurotransmitter release as an initiating event can lead to inclusions pathology and neuronal demise. An answer to this question may provide insights regarding the early steps that lead to neurodegeneration, and that could further identify targets for preventative intervention. For example, development of drugs that inhibit α-synuclein actions at the synapse could be a potentially effective treatment for Parkinson disease.
References:
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997 Jun 27;276(5321):2045-7. PubMed.
Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998 Feb;18(2):106-8. PubMed.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003 Oct 31;302(5646):841. PubMed.
Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004 Feb;55(2):164-73. PubMed.
Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005 Nov 4;123(3):383-96. PubMed.
Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006 Nov 15;26(46):11915-22. PubMed.
Rockenstein E, Schwach G, Ingolic E, Adame A, Crews L, Mante M, Pfragner R, Schreiner E, Windisch M, Masliah E. Lysosomal pathology associated with alpha-synuclein accumulation in transgenic models using an eGFP fusion protein. J Neurosci Res. 2005 Apr 15;80(2):247-59. PubMed.
Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001 Nov 2;294(5544):1117-22. PubMed.
Nonet ML, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci. 1998 Jan 1;18(1):70-80. PubMed.
Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. J Neurosci. 1998 Mar 15;18(6):2028-39. PubMed.
Make a Comment
To make a comment you must login or register.