PET Ligand Lights Up AAIC, May Detect Synapse Loss in AD
Quick Links
Among Alzheimer’s disease pathologies, synaptic loss correlates best with symptoms, but until now researchers have had no way to measure it in living people. That may be changing, with a new PET ligand that binds synaptic vesicle glycoprotein 2A (SV2A). At the Alzheimer’s Imaging Consortium symposium on the day before this year’s Alzheimer’s Association International Conference, July 22–26 in Chicago, Richard Carson of Yale University, New Haven, Connecticut, reported that the probe, UCB-J, bound significantly less in the hippocampi of people with Alzheimer’s disease than in controls. Poor retention of the ligand correlated with flagging memory, he showed. The work was published in the July 16 JAMA Neurology, and Alzforum previously reported some of the data when Christopher van Dyck, also from Yale, presented last fall at the Clinical Trials on Alzheimer Disease meeting in Boston (Dec 2017 conference news).
At AAIC, multiple attendees told Alzforum the work was a highlight of the imaging consortium session. “PET measurements of presynaptic and postsynaptic density would greatly advance the scientific study of Alzheimer’s disease and related disorders," said Eric Reiman, Banner Alzheimer’s Institute in Phoenix. "The Yale group has developed this promising 11C-labeled ligand to assess presynaptic density. They have begun to develop an 18F-labeled ligand that would advance its use by other centers, and they have begun to test these radioligands in rigorous and thoughtful ways," he wrote in an email to Alzforum (see complete comment below).
Rik Ossenkoppele, Vrije University, Amsterdam, called the work impressive and important. “The availability of a PET tracer that allows detection of structural synaptic alterations has important potential implications for clinical studies,” he wrote (see comment below).
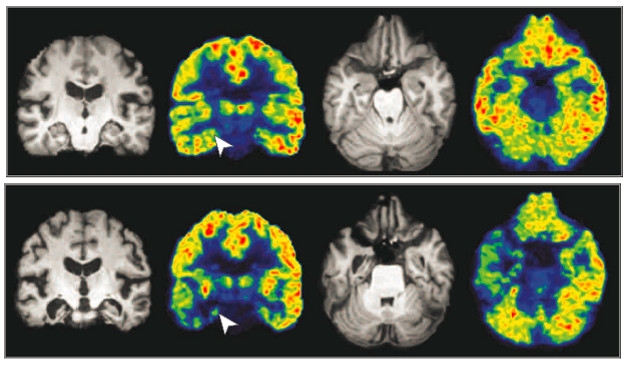
Synapses in Decline? Coronal and axial pseudocolor maps show intensity of UCB-J binding in one cognitively normal person (top) and one AD patient (bottom). Arrows mark the right hippocampi in the coronal view. Reference MRI images are gray. [Image © (2018) American Medical Association. All rights reserved.]
Others were more reserved, asking if the probe truly bound synapses or some other vesicle fraction in the brain. Carson said that has yet to be worked out. “We need to understand the relationship between vesicles and synapses, and how it is affected by vesicle metabolism, including exocytosis and recycling, even synaptic activity,” he said. One audience member questioned if the distribution of synapses in the human brain maps to UCB-J binding. Carson replied that electron microscopy and other work to address this is ongoing. Some were concerned that the binding pattern varies from that seen with FDG, a PET ligand that measures glucose metabolism and is widely considered a surrogate for synaptic activity.
Still, the overall mood at AAIC was that this ligand could provide useful information about the brain. Developed by Carson, 11C-UCB-J binds with high affinity and specificity to SV2A, a universal marker of presynaptic vesicles. SV2A distribution closely matches that of synaptophysin, which is commonly used as a marker for synapses in postmortem human tissue. Previously, Carson and colleagues reported that it detected synaptic loss around focal seizures in people with epilepsy (Jul 2016 news).
In the AD study, Ming-Kai Chen, who works in Carson’s group and is first author on the JAMA Neurology paper, measured brain binding of 11C-UCB-J in 10 people who had mild cognitive impairment or dementia and who had tested positive on amyloid PET scans. He compared them to 11 cognitively normal, amyloid-negative, age-matched counterparts. The investigators focused their attention on the hippocampus, where they expected early synapse loss due to the degeneration of projections from the entorhinal cortex. They calculated relative binding of UCB-J using centrum semiovale white matter as a reference region, because it poorly binds the ligand.
The Alzheimer’s patients bound 41 percent less tracer in the hippocampus than did controls. That difference held even after correction for hippocampal atrophy. Chen also found lower binding in the entorhinal cortex, but that was explained by loss of tissue volume.
Postmortem examination of AD brains reveals widespread synaptic loss in the cortex, yet in this study, PET detected no difference between cortical binding of 11C-UCB-J in patient and control cortices. Why? The authors speculate the study was too small, or that most of the AD patients were at an early disease stage and had not yet lost cortical synapses. Another possibility is that remaining synapses had grown to compensate for those that had withered. This phenomenon, called synaptic hypertrophism, has been documented in mild AD (DeKosky and Scheff, 1990). Ossenkoppele wondered if the patient-selection criteria, which required poor scores on an episodic memory test, had enriched for people with primarily limbic pathology, i.e., hippocampal atrophy. Chen told Alzforum they plan follow-up studies, including postmortem analyses, with more patients in later stages of disease.
In an editorial accompanying the paper, Elizabeth Mormino of Stanford University, Palo Alto, California, and William Jagust, of University of California, Berkeley, raised the FDG issue. They point out that the decrease in UCB-J binding observed only in the hippocampi of people with AD differs dramatically from the more widespread cortical hypometabolism detected using FDG-PET. “This is surprising, because although hippocampal synaptic loss is severe, other brain regions, such as the frontal cortex, entorhinal cortex, temporal cortex, and cingulate cortex, have been reported to show a similarly high synaptic loss in postmortem studies that may even predominate in the presynaptic terminals measured with an [sic] synaptic vesicle glycoprotein 2A ligand,” they write.
Others have puzzled over this FDG/UCB-J dichotomy, as well. “Since FDG PET has been suggested to provide information about the density, activity, and metabolism of terminal neuronal fields and/or peri-synaptic glial cells, it will be important to understand the biological basis for these differences,” noted Reiman. Victor Villemagne, University of Melbourne, Australia, wondered why UCB-J did not detect synapse loss in cortical areas. “Is function (FDG) more sensitive than synaptic density?’” he asked (see comment below).
Chen and colleagues did detect less blood flow in some cortical regions of the MCI/dementia patients compared with controls. These were the regions where FDG-PET wanes in people with AD, but this blood flow reduction seemed to have no effect on UCB-J binding. Chen said this underscores that the two probes do not measure the same thing—where the FDG-PET signal provides a composite of glucose uptake by pre- and postsynaptic neurons and nearby glia, UCB-J provides a snapshot of synaptic structure and integrity. For this reason, he said, “We do not think that UCB-J-PET will replace FDG-PET, but instead that the two will complement each other.”
In a poster at AAIC, co-author Adam Mecca presented results from a preliminary analysis where he directly compared UCB-J and FDG-PET in nine people with MCI or AD, and 11 controls. He reported the expected pattern of FGD-PET hypometabolism in multiple cortical regions in the patients, and reduction of UCB-J binding in the hippocampus, but again no other regions. The investigators found a correlation between FDG-PET and UCB-J-PET signals in the hippocampus across both patients and healthy controls.
Other labs plan to test the probe. Bradley Christian, University of Wisconsin, Madison, said his group synthesized the tracer in-house, and completed animal testing before applying for FDA approval for human studies.
Carson and colleagues are developing 18F probes, which would expand use beyond centers that can make their own supply of the short-lived 11C version. Here things get tricky. UCB-J contains three fluorines. “We have labelled it with F18, but the chemistry is terribly difficult,” Carson admitted. Instead, the researchers have turned to making derivatives that contain a single fluorine. Two, called SDM8 and SDM2, look promising, said Carson. He said SDM8 has nearly identical kinetics to UCB-J, while SDM2 appears to enter and leave the brain more rapidly and might be useful for short scans. Both are being tested in human studies, he said. Independently, researchers in Belgium are working on 18F SV2A ligands (Bahri et al., 2017), as are investigators at Invicro, an imaging company based in Boston. Chen predicts the 18F probes will become more widely available in the next year or two, allowing more groups to experiment with the tracer.
UCB-J is being used in small pilot trial of CT1812, a sigma-2 receptor antagonist that purportedly protects against Aβ synaptotoxicity. The 21 patients will undergo PET scans at baseline and after six months’ treatment with either placebo or the drug. Chen said several volunteers have already completed their baseline scans.—Pat McCaffrey and Tom Fagan
References
News Citations
- At CTAD, Tau PET Emerges as Favored Outcome Biomarker for Trials
- Next Up for Human Brain Imaging: Synaptic Density?
Therapeutics Citations
Paper Citations
- Dekosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990 May;27(5):457-64. PubMed.
External Citations
Further Reading
Papers
- Finnema SJ, Nabulsi NB, Mercier J, Lin SF, Chen MK, Matuskey D, Gallezot JD, Henry S, Hannestad J, Huang Y, Carson RE. Kinetic evaluation and test-retest reproducibility of [11C]UCB-J, a novel radioligand for positron emission tomography imaging of synaptic vesicle glycoprotein 2A in humans. J Cereb Blood Flow Metab. 2017 Jan 1;:271678X17724947. PubMed.
Primary Papers
- Chen MK, Mecca AP, Naganawa M, Finnema SJ, Toyonaga T, Lin SF, Najafzadeh S, Ropchan J, Lu Y, McDonald JW, Michalak HR, Nabulsi NB, Arnsten AF, Huang Y, Carson RE, van Dyck CH. Assessing Synaptic Density in Alzheimer Disease With Synaptic Vesicle Glycoprotein 2A Positron Emission Tomographic Imaging. JAMA Neurol. 2018 Oct 1;75(10):1215-1224. PubMed.
- Mormino EC, Jagust WJ. A New Tool for Clinical Neuroscience-Synaptic Imaging. JAMA Neurol. 2018 Jul 16; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Arizona Alzheimer's Consortium
PET measurements of presynaptic and postsynaptic density would greatly advance the scientific study of Alzheimer’s disease and related disorders. The Yale group has developed this promising 11C-labeled ligand to assess presynaptic density. They have begun to develop an 18F-labeled ligand that would advance its use by other centers, and they have developed and tested these radioligands in extremely rigorous and thoughtful ways. The group has also found surprising but potentially informative differences between the pattern of UCB-J and FDG PET reductions in clinically affected persons with AD. Since FDG PET has been suggested to provide information about the density, activity, and metabolism of terminal neuronal fields and/or peri-synaptic glial cells, it will be important to understand the biological basis for these differences. Additional studies are needed to clarify the role of this promising PET method to the detection and tracking of AD-related synaptic loss and in the evaluation of putative disease-modifying treatments.
VU University Medical Center
This is very impressive and important work. I commend the authors for their well-executed study and the PET methodology is truly excellent. The authors demonstrated for the first time reduced synaptic density in the hippocampi of living persons with early stage Alzheimer’s disease compared to healthy controls. The availability of a PET tracer like 11C]UCB-J that allows detection of structural synaptic alterations has important potential implications for use in clinical (synaptic loss is intimately linked to cognitive impairment and could be used to improve diagnosis and prognosis), investigational (combining the three key elements of AD pathophysiology, i.e., Aβ, tau, and synaptic density), or clinical trial (surrogate outcome measure for therapies tailored to rescue/restore synapses) settings. However, my excitement about 11C]UCB-J PET was slightly lowered by two observations.
1. The spatial pattern of decreased 11C]UCB-J binding was restricted to the hippocampus and entorhinal cortex, and there were no significant differences between patients and controls in neocortical areas. Also, there was quite substantial overlap in hippocampal 11C]UCB-J binding between AD patients and controls. Although very little postmortem data exists on synaptic density in early AD, it seems unlikely that the ongoing disease process (i.e., accumulation of Aβ and presumably tau proteins) for ~10–15 years has no consequences for the vitality of neocortical synapses. The authors and accompanying editorial by Mormino and Jagust offer several explanations, including limited statistical power due to the relatively small sample size, the mild disease stage of patients and synaptic hypertrophy as a potential compensatory mechanism. Another explanation could be that the AD patients were selected for an amnestic-predominant phenotype based on a memory test. This category of patients is overrepresented in the “limbic-predominant” subtype of AD (Murray et al., 2011; Whitwell et al., 2012), which has been associated with spatially more confined patterns of neurofibrillary tangle pathology and brain atrophy in limbic regions. Future work including clinically more advanced patients and non-amnestic variants of AD are needed to better understand the lack of neocortical 11C]UCB-J signal.
2. The associations between 11C]UCB-J binding and cognition were less strong than would be expected from neuropathological data. In this proof-of-concept study, the authors show significant associations when combining controls and AD patients, but within the AD group there seems to be no relationship between hippocampal 11C]UCB-J binding and memory impairment, as equivalent memory scores were observed in AD patients with 11C]UCB-J binding potentials around 0.5 and 1.5 (Figure 2B in the paper). This also needs further investigation in larger and more heterogeneous samples of AD patients, preferentially in longitudinal studies.
Overall, this study adds a new and promising PET technique to the existing set of biomarkers that are currently used to study AD. I am looking forward to studies investigating longitudinal changes in synaptic densities and its associations with clinical progression and structural, functional, and pathophysiological biomarkers of AD. However, based on the presented spatially confined 11C]UCB-J binding pattern in a neurodegenerative disease with such a clear neural signature, I have some reservations about the usefulness of 11C]UCB-J PET for studying psychiatric disorders such as depression, autism, or schizophrenia, in which the distinction with normal controls is expected to be much smaller.
Austin Hospital
Imaging synaptic density opens a new avenue to assess neurological conditions, in particular neurodegenerative conditions such as Alzheimer’s disease where a synaptopathy, either as a consequence of the toxic effects of aggregated proteins and/or other factors, is at the center of its pathophysiology.
11C-UCB-J, or its F-18 derivative, will need to be validated. On the other hand, the discrepancy of the perfusion as well as persistent significance in the hippocampus even after partial volume correction, indicates 11C-UCB-J measures a different variable than FDG. Also, as Chen and colleagues point out, it is not clear if there are compensatory mechanisms to maintain the number of presynaptic vesicles, or a compensatory sprouting in nigrostriatal pathways as observed in Parkinson’s disease.
The association with cognitive performance—where participants from all clinical categories were pooled together—moves in the right direction, with lower synaptic density associated with worse cognitive performance. When we look at the discrepancy in the extent and pattern with FDG and atrophy we need to assess where the synaptopathy is worst. Is it pre- or postsynaptic? On the postsynaptic, de novo-formed tau aggregates inside dendrites might lead to their shrinking. And the relationship with tau has other connotations, too. Hyperphosphorylation of tau leads to it detaching from microtubules, disrupting axonal transport, and lowering the transport of proteins and organelles to the presynaptic terminal.
The data also raise other questions: Why is there no decrease of synaptic density in other cortical areas, especially those known to be associated with affected cognitive domains in AD? Is function (FDG) more sensitive than synaptic density? The data also generate a wish list of areas to be explored: How does synaptic density change in early onset AD, with marked parietal atrophy? In atypical presentations of AD? In hippocampal sclerosis? In autosomal-dominant cases?
We need to emphasize the importance of being able to assess synaptic density in vivo both in cross-sectional and longitudinal fashion that, similar to amyloid imaging, will translate in better patient selection, assessment of target engagement, and efficacy at trial of therapies aimed at preventing neurodegeneration.
Make a Comment
To make a comment you must login or register.