CONFERENCE COVERAGE SERIES
EMBO | EMBL Symposium: Mechanisms of Neurodegeneration
Heidelberg, Germany
14 – 17 June 2017

At Heidelberg meeting, scientists shared and debated new data on microglial function.
CONFERENCE COVERAGE SERIES
Heidelberg, Germany
14 – 17 June 2017
At Heidelberg meeting, scientists shared and debated new data on microglial function.
New insights into microglial biology and function created quite the buzz at Mechanisms of Neurodegeneration, an EMBO/EMBL symposium held June 14-17. Organized by Karin Dumstrei, Todd Golde, Christian Haass, and Bart De Strooper, the second iteration of this meeting drew about 200 researchers to the EMBO training and conference center in Heidelberg, Germany. Topics ran the gamut from old and knotty questions such as how γ-secretase works, to hot topics like microglial pruning of synapses, to completely unexpected ideas, i.e., can microglia seed plaques?
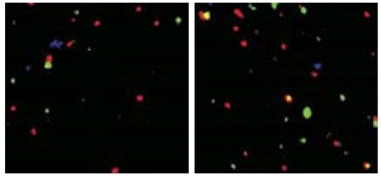
Hippocampal CA3 tissue from APPPS1 mice (left) has less presynaptic Vglut2 (green) and postsynaptic GluR1 (red) than CA3 in APPPS1 C3 knockouts (right). [Courtesy of Shi et al., 2017; Science Translational Medicine/AAAS.]
Beth Stevens, Children’s Hospital, Boston, set the tone for the meeting by reviewing the role of microglia in Alzheimer’s and Huntington’s diseases. Over the last few years, seminal work has changed how the field views microglia in neurodegeneration. Scientists discovered how these immune cells resume a job they once had early in brain development and then retired; that is, to prune synapses. Central to this lies the complement signaling cascade, kick-started by complement C1q and fueled by complement C3, which flags structures that are to be engulfed by phagocytic cells. Stevens outlined recently published data from her and Cynthia Lemere’s labs at Brigham and Women’s Hospital, in which knocking out C3 protected APP/PS1 mice from learning and memory loss, albeit at the cost of much more Aβ than usually accrues in this AD model (see Jun 2017 news).
In her talk in Heidelberg, Stevens extended this concept to Huntington’s disease. She reported that C1q and C3 are upregulated in mice expressing mutant human huntingtin with an expanded polyglutamine repeat, and that the animals retain more synapses after chronic treatment with an anti-C1q antibody. “That they find the exact same thing going on in parallel in two different models strongly suggests that we have a common mechanism for synaptic pruning in neurodegenerative diseases,” said Haass, from the German Center for Neurodegenerative Diseases (DZNE) in Munich. “For me, this was one of the highlights of the meeting and other people seemed to think so, as well,” Haass told Alzforum. Stevens said she is now testing whether complement deficiency affects huntingtin pathology and motor function in HD mice.
Stevens’ work supports the idea that phagocytic microglia simultaneously clear amyloid plaques and prune synapses. In contrast to that, might these cells also contribute to plaque formation? Michael Heneka from the DZNE in Bonn showed data on protein conglomerates called ASC specks. Short for “apoptosis-associated speck-like protein containing a caspase-recruitment domain,” ASC aggregates when activated by the inflammasome, forming bundles that appear as specks under the light microscope. ASC specks activate caspase-1, which in turn activates inflammatory cytokines such as interleukin 1β and IL-18, powering an inflammatory cascade. Released by microglia, ASC specks can be taken up by nearby cells, unleashing the same inflammatory cascade in them (Jun 2014 news). Heneka previously reported that microglia in the frontal cortices of AD patients spew out ASC specks. In keeping with this, he found that silencing the NLRP3 inflammasome that activates ASC can protect APP/PS1 mice from learning and memory deficits (Dec 2012 news).
What then, causes the release of ASC specks in AD? In Heidelberg, Heneka said he initially suspected Aβ, but experiments done in his lab to prove this hunch did not pan out. That’s because it turned out that ASC specks have a voracious affinity for binding Aβ. While that appetite complicated the experiments, it led Heneka to a more important idea—that ASC specks help seed and spread Aβ deposits in the brain. With a movie of microglia under the microscope, Heneka showed that Aβ latches onto ASC specks like iron filings to a magnet. He went on to describe, in a series of in vitro biochemical and cell biology experiments, which regions of the ASC structure bind Aβ, how ASC can pull Aβ out of solution by gravity, and that ASC specks seed formation of Aβ oligomers.
Does this matter in vivo? Heneka reported that ASC always turns up in Aβ plaques in mouse models of AD, and crossing those models with ASC knockouts dramatically reduces levels of soluble and insoluble Aβ in APP/PS1 mice, while also rescuing their learning and memory deficits. Haass called this work a hot new topic. “That Michael finds the core of Aβ plaques always have ASC specks is super surprising,” Haass said.
All told, Heneka’s data suggest that ASC specks could play a role in the seeding and spreading of plaques. He tested this by injecting Aβ-containing lysates into mouse brains as done in now-classic Aβ seeding experiments by Mathias Jucker and Lary Walker (see, for example, Sep 2006 news on Meyer-Luehmann et al., 2006). When Heneka injected three-month-old APP/PS1 mice with Aβ it increased the amount of deposits around the brain five months later, as seen on serial brain sections. However, in APP/PS1 x ASC knockouts, injected lysates caused no ramp-up in plaque deposition. ELISA assays of total brain Aβ backed this up.
Haass considered the data compelling. “I have found that Aβ injected into the brain has never really seeded well—you have to flood the brain with peptide. So there is something missing in regular seeds [that’s needed for full propagation], and the ASC speck may be it,” he told Alzforum.
What about ASC specks in patients? Preliminary evidence suggests they play a role there, too. Heneka and colleagues have immunoprecipitated the complex from samples of postmortem brain tissue; in the case of AD and mild cognitive impairment, this pulls down Aβ as well. They detected no Aβ in pulldowns from normal age-matched controls or from people who had had frontotemporal dementia or corticobasal degeneration. In fact, Heneka showed an image of a brain section from an AD patient that displayed what he called “microglial Armageddon,” with an abundance of Iba1-expressing microglia littered with adjacent ASC specks. Side by side with early drawings of microgliosis penned by Alois Alzheimer himself, the two looked remarkably similar.
Scientists at the meeting avidly discussed Heneka’s presentation. Marco Colonna, Washington University, St. Louis, asked if microglia digest the ASC speck/Aβ complexes—au contraire, Heneka said they are extremely stable and resist degradation. Stevens wondered if astrocytes play a role in this ASC pathology—Heneka plans to investigate that. He did note that astrocytes appear to atrophy more at sites of ASC release, but since Aβ pathology is rampant there, too, Heneka can’t be sure which might be driving astrocyte cell death.
Others questioned the role of ASC specks in AD pathology. Konrad Beyreuther from the University of Heidelberg was perplexed that ASC specks reportedly have higher affinity for Aβ40 than for Aβ42. “Why then do we only see Aβ42 in human plaques?” he asked. Heneka had no immediate answer for that, but others speculated that Aβ42 might be needed to template misfolding and aggregation of more Aβ on the ASC speck core.
Other scientists described new findings on the homeostasis of microglia. Researchers led by Frank Heppner at Charité – Universitätsmedizin Berlin had previously reported that myeloid cells infiltrating the brain were strangely uninterested in mopping up amyloid plaques (Apr 2015 conference news and Prokop et al., 2015). Why might that be? In Heidelberg, Heppner posited that something in the AD brain suppresses the cells. He reported that peripheral myeloid cells that infiltrate the brain in normal mice sport more dynamic soma and cellular processes than do myeloid cells infiltrating the brain of an AD mouse model. Researchers in his lab found that an interleukin cascade might be silencing the cells in the disease state. Knocking down p40, a subunit of both IL12 and IL23, reduced plaque pathology in APP/PS1 transgenic mice (Nov 2012 news). Heppner said that p40 and IL12 levels climb in AD patients, while a polymorphism in the IL23 receptor has been linked to AD in Han Chinese (Hu et al., 2012; Sudduth et al., 2012; Liu et al., 2014). Heppner now has data to suggest that the cognate receptors for IL12/23 in the brain lie on astrocytes, suggesting that a cytokine-mediated cross-talk between these cells and microglia temper the latter. To test that, researchers in his lab have conditionally knocked out the IL12 and IL23 receptors in astrocytes and crossed those animals with APP/PS1 mice. Time will tell whether the offspring are also protected from amyloidosis like the IL12/23 knockouts.
Heppner’s data might not only shed light on microglial deficiencies in AD, but could have implications for therapy as well. He reported that Aβ immunotherapy is ineffective in AD mouse models whose microglia are depleted; indeed, the more microglia there are, the more efficacious the antibodies. Heppner also reported a proof of the peripheral sink hypothesis, which posits that mopping up Aβ in the blood speeds up its clearance from the brain. He generated mice that make IgM antibodies against Aβ in the periphery and showed that they have more Aβ in the blood but one-third less plaque burden than controls. Haass called these experiments important. “This data fit nicely with our report that TREM2 deficiency reduces antibody-induced clearance of amyloid,” he said. “This confirms that we really need microglia for immunotherapy to work.”
This meeting featured a smidgeon of new data about TREM2, when Colonna confirmed and extended findings from the Haass lab that this microglial receptor plays major roles in microglial homeostasis and reported the new finding that TREM2 functions in the regulation of autophagy, as well. Evidence for the latter came from examining TREM2-negative microglia in 5xFAD mice. The cells were packed with multilamellar vesicles, suggestive of increased autophagic clearance, and they had accumulated lipidated LC3, an autophagosomal marker. In keeping with this, Colonna reported weaker signaling through mammalian target of rapamycin (mTOR) in TREM2-negative microglia. A central regulator of autophagy, mTOR coordinates growth and nutrient signals in cells. Colonna found that TREM2-negative microglia were unable to muster normal levels of many of the intermediaries generated by classic metabolic pathways. These include glycolysis, the pentose phosphate shunt, and the tricarboxylic acid cycle.
All told, Colonna concluded that the absence of TREM2 signaling suppresses normal metabolic function in microglia, which then try to compensate by increasing autophagy. “Temporarily, that counterbalance might be good, but eventually the cells become apoptotic and die,” he said.—Tom Fagan
In Alzheimer’s disease, the axonal protein tau accumulates aberrantly in dendritic spines, where it contributes to excitotoxicity. What causes this mislocalization? Previous studies have linked hyperphosphorylation or overexpression of the protein to its accumulation in dendrites. At the Mechanisms of Neurodegeneration conference held June 14-17 in Heidelberg, Germany, Jürgen Götz of the University of Queensland in Brisbane, Australia, proposed a different explanation. Tau levels spike in dendrites because the protein is synthesized on the spot, he said. Aβ oligomers trigger this local production through a pathway that involves the kinase Fyn. “This alternative mechanism may explain the somatodendritic accumulation of tau induced by Aβ,” Götz told Alzforum. He believes the findings support therapeutic strategies that target tau production, rather than phosphorylation.
Other speakers in Heidelberg focused on specific ways for getting rid of dendritic tau. Karen Duff of Columbia University, New York, discussed how to use the neuropeptide PACAP to activate the proteasome in dendrites, thus chewing up unwanted tau. This approach might help prevent the spreading of tau from cell to cell early in disease, she noted. Researchers in attendance found the idea intriguing, peppering her with questions. Lennart Mucke of the Gladstone Institute of Neurological Disease, San Francisco, called the talk impressive. “This data is in line with other studies suggesting that enhancing the proteasomal degradation of tau could be of therapeutic benefit in AD and other tauopathies,” he told Alzforum.
In the last few years, several studies have tied dendritic tau to excitotoxicity and synaptic damage (see Sep 2010 news; Jan 2011 news). Götz previously worked out one mechanism for this, finding that tau escorts Fyn to the synapse, where the kinase then mediates excitotoxic signaling by Aβ (Jul 2010 conference news).
These results led Götz and Chuanzhou Li at Queensland to further study how tau and Fyn interact. They transfected both genes into HEK293 cells, and noticed a curious thing: Transfection of tau alone produced but a faint signal from the exogenous protein, whereas co-transfection of both genes boosted tau expression roughly 30-fold. Investigating this, they found that the increase was due to greater translation of tau, rather than transcription. The interaction was specific for tau and Fyn, as Fyn did not jack up translation of any other proteins they examined.
To dissect the mechanism, the researchers examined various components of the translational machinery, and manipulated them with activators and inhibitors. They delineated a pathway that runs from Fyn kinase through ERK and S6 kinase to pump up tau translation. Turning to mouse hippocampal primary neurons, they found that this same pathway acted in these cells, and that it could be kicked off by adding synthetic oligomeric Aβ to the culture. Because the kinases in this pathway also phosphorylate tau, the excess protein becomes hyperphosphorylated, Götz noted.
Does this happen in vivo? The researchers found that neurons cultured from Fyn knockout mice made 60 percent less tau, while animals with constitutively active Fyn had excess tau. In APP23 mice, Fyn, ERK, and S6 were all highly activated. Injecting Aβ oligomers into wild-type mice also activated these kinases, and raised tau levels in the affected neurons, Götz said. Meanwhile, Fyn inhibitors turned down production induced by Aβ.
To find out where in the cell this tau translation was occurring, Götz and Li used a proximity ligation assay that allowed them to label newly synthesized tau. They found the new protein only in the somatodendritic compartment, not in axons. The researchers fractionated lysates from APP23 brain and confirmed high tau levels in the cytosolic and synaptosomal fractions. By comparing older and younger mice, the researchers concluded that tau rises first in the soma, and then later spreads to dendrites.
The findings intrigued Christian Haass, Ludwig Maximilians University in Munich, who pointed out that cerebrospinal fluid tau levels rise steeply in AD, but not in frontotemporal dementia, even though tau pathology accumulates in many forms of that disease (Hampel et al., 2004; Olsson et al., 2005). Perhaps this discrepancy occurs because the FTD brain does not contain Aβ oligomers, and therefore does not turn up tau production, Haass suggested.
What are the consequences of local tau synthesis? Because dendritic tau can corral more Fyn, leading to excitotoxicity, while Fyn further boosts tau production, the pathways may form a vicious circle, Götz noted. He believes the data support therapeutic strategies that lower total tau levels. The tau immunotherapy field has been moving in this direction already. Other approaches would be to silence tau expression using antisense oligonucleotides or microRNA, Götz suggested. Targeting ERK or S6 kinase would not be an option, because these enzymes affect so many cellular processes, but Fyn might be a viable target for turning down tau synthesis, he added. Fyn does play a key role in myelination, but this process is finished in adults.
“The findings shed new light on well-established links among Aβ, Fyn, and tau, and further solidify the rationale for inhibiting Fyn and related signaling pathways in AD,” Mucke wrote to Alzforum. However, he noted that his own group has not found elevated tau levels in the J20 mouse line, which, like APP23, expresses human mutant APP. “Additional studies will likely resolve whether these differences are due to the age at which the mice were analyzed, the methods used, or other variables,” he wrote.
For her part, Duff’s talk covered a different approach for lowering tau. She had previously found that tau aggregates clog the proteasome, causing tau clearance to grind to a halt. Raising cyclic AMP levels counteracts this by switching on protein kinase A, which in turn activates the proteasome to mop up aggregates. Drugs that boost cAMP levels, such as rolipram and cilostazol, lowered tau levels and improved learning and memory when given to young rTg4510 mice (Dec 2015 news).
In Heidelberg, Duff described a more selective way to locally raise cAMP in dendrites. The neuropeptide PACAP activates the G-protein coupled receptor, PAC1, which elevates cAMP levels and is expressed in neuronal cell bodies and dendrites but not axons (Joo et al., 2004). Using an osmotic pump to administer a constant trickle of PACAP to rTg4510 mice lowered tau specifically in the postsynaptic fraction, not in presynapses or total cell lysate, Duff said. Proteasome activity ramped up, and memory in the Morris water maze and novel object recognition test improved in the treated mice.
The experiment acts as a proof of concept for this approach, Duff suggested. She believes it may be possible to target other receptors, such as muscarinic or adrenergic ones, to affect cAMP. Activating the proteasome could also help clear other aggregating proteins, for example α-synuclein or FUS, she added. In future work, she will also look at whether this strategy limits the spread of pathologic tau. The approach does not work for mice at advanced stages of diseases, she noted. “Once the proteasome is dead, you cannot rescue it,” she said. Götz noted that data suggest PACAP and the PAC1 receptor play a role in post-traumatic stress disorder and psychiatric conditions. “There may be a broader application of this therapeutic strategy,” he wrote to Alzforum.
Bart De Strooper of the U.K. Dementia Research Center at UCL urged caution, noting that the full picture may be more complicated since G-protein coupled receptors have been linked to various aspects of amyloid pathology. The PAC1 receptor, for example, also regulates ADAM10, which is responsible for the α-cleavage of APP, De Strooper pointed out, adding that there is a web of complicated GPCR systems. Duff acknowledged this, but suggested that specific pharmacological interventions might be worked out.—Madolyn Bowman Rogers
No Available Comments
The Alzheimer’s disease proteome has remained mostly a mystery. At the EMBO/EMBL Symposium on Mechanisms of Neurodegeneration, held June 14-17 in Heidelberg, Germany, Nick Seyfried described one of the first major attempts to chart this unexplored territory. Working with Allan Levey at Emory University in Atlanta, Seyfried has analyzed levels of more than 10,000 proteins from about 40 control, Alzheimer's disease (AD) and Parkinson's disease (PD) patients and controls. He used bioinformatic network analysis to uncover suites of proteins that correlated with Aβ and tau pathology. This work, he hopes, will help identify drivers of disease pathology or new biomarkers. “The Emory team is the only one doing large-scale AD proteomics; they are pushing the limits of what you can do with mass spectrometry,” said meeting co-organizer Todd Golde from the University of Florida, Gainesville. “To have a centralized repository of this size and scope will be incredibly valuable to the community,” Golde said.
The project is funded by the Alzheimer’s disease initiative of the Accelerating Medicines Partnership, a joint venture between the National Institutes of Health, the Food and Drug Administration, and 10 biopharmaceutical companies and multiple nonprofits. The AMP has tasked a consortium of academic teams to build and openly share large, complex AD data sets (see Feb 2014 news).

A protein module associated with inflammation, enriched in astro- and microglia markers (red), positively correlates with AD. A module enriched in neuronal markers (blue) negatively correlates with AD. “Hub Proteins” are those whose expression levels correlate most tightly in each module. [Cell Systems, Seyfried et al., 2017.]
Seyfried expects proteomics will complement existing transcriptomics data. Because translation is a regulated process, RNA transcript levels don’t always mirror the levels of the proteins they encode. Indeed, in isolated mouse brain cells, that correlation reaches 0.47 at most (Sharma et al., 2015). In addition, proteomics analyses can identify post-translational modifications, protein fragments such as soluble TREM2 or soluble APP, and proteins that find their way into the brain from the periphery. For example, Golde noted that several complement proteins are found at high levels in the cerebrospinal fluid in AD patients and probably accumulate in their brains. “Because AD is a proteinopathy, proteomics is as important, if not more so, than transcriptomics,” Golde said.
The Emory researchers have set up two analytical pipelines. One is quick, relatively inexpensive, and creates estimates of the most abundant proteins in high-throughput fashion. The second is slower and pricier, but dives more deeply into the proteome pool.
Seyfried showed how he used the quicker approach, known as label-free, single-shot proteomics, to quantify roughly 3,000 proteins in each of about 800 tissue samples. The hope is that these robust signatures could identify potential biomarkers. With its ability to process many samples at once, single-shot proteomics also facilitates protein network analyses, which rely on many replications for accuracy.
The second approach labels peptides with multiple chemical tags allowing the least abundant proteins to be retrieved and quantified. Seyfried said his team can now track about 10,000 proteins, mapping to more than 10,200 expressed genes covered in recent transcriptome efforts. Researchers called this a significant feat, considering that proteins cannot be amplified the way mRNA can.
In his talk, Seyfried covered data from the high-throughput technique (Seyfried et al., 2017). He has studied postmortem samples from the dorsolateral prefrontal cortex and the precuneus of 15 controls, 15 asymptomatic people who had AD as judged by postmortem CERAD and Braak staging scores, and 20 people who had had an AD diagnosis before death. All came from the Baltimore Longitudinal Study of Aging. The researchers identified 5,130 different proteins, but focused on 2,735 that were found in at least 90 percent of the samples from each brain region. The abundance of two peptides corresponding to residues 6-16 and 17-28 of Aβ strongly correlated with CERAD scores of amyloid pathology, lending confidence to the approach.
To better assess differences between AD and control samples, the researchers turned to weighted co-expression network analysis, following the lead of previous transcriptomics studies (Zhang and Hovarth, 2005; Miller et al., 2008; Miller et al., 2013). “This analysis correlates expression of proteins in an agnostic manner, without using information about the neuropathology of the samples,” said Seyfried. First it compares each protein, pairwise with others in each sample, and then clusters them into highly correlated groups, or modules. Seyfried found 16 such modules in the BLSA samples. Because the results from the prefrontal and precuneus regions were highly similar, he combined the data to gain power.
The largest module comprised mostly neuron-specific proteins, such as those involved in synaptic transmission and neurite biology. Other modules reflected basic cellular functions, such as protein folding, or proteins found in particular organelles, such as mitochondria. Modules enriched in proteins specific to astrocytes, oligodendrocytes, and microglia emerged, judging by similarities to proteomes from isolated mouse brain cells. “We could infer from the modules what might be happening in specific cells without having to isolate them,” said Seyfried.
As expected from transcriptome studies, the researchers saw the largest AD-associated changes in modules enriched for cell type-specific proteins. However, the correlation between the calculated “eigenprotein,” aka the first principal component of a module, and CERAD and Braak staging differed for glia and neurons. For astrocyte- and microglia-enriched modules, the eigenprotein correlation strengthened as pathology worsened, whereas for neurons, the correlation weakened (see image above). This is in line with transcriptomic data, which found increases in modules associated with immune responses, in particular microglia, in early AD.
Changes in functional modules correlated with diagnosis. Levels of proteins involved in inflammation and apoptosis were higher in AD than controls. Curiously, apoptosis hub protein levels were slightly higher in controls than in asymptomatic AD patients, even though the latter have accumulated amyloid plaques. Seyfried wondered if some protective mechanism may be at play in individuals who have amyloid in the brain but are still cognitively normal.
While cell-specific proteomic modules overlapped with previously identified RNA modules, the overall overlap of protein and RNA, including non-cell-specific modules such as apoptotic or mitochondrial, was only 39 percent. Seyfried thinks this may reflect, in part, the distance between soma and axons. Protein changes in the latter would be picked up by proteomics, but not by transcriptomics if the transcripts stay near the nucleus. Also, protein from the periphery may build up to a greater extent in AD and aging brains as a result of disruption of the blood-brain barrier, he suggested
Are these changes cause or consequence of AD? Eric Reiman of the Banner Alzheimer’s Institute in Phoenix suspects it’s a bit of both. Working with Dietrich Stephan, now of the University of Pittsburgh but previously at the Translational Genomics Research Institute in Phoenix, Reiman had found that transcripts encoding some of the neuronal and mitochondrial proteins Seyfried identified were also downregulated (Liang et al., 2008). Seyfried believes proteomic network analysis will help reveal upstream triggers of disease, but Golde cautioned that bioinformatics will go only so far. “Deconvoluting causality is asking a lot from these data. This type of data set gives us a large number of samples as reference points. We’re not there yet with respect to causality,” he said.
Is network analysis enough to tease apart the roles of different cells in AD? Bart De Strooper of the U.K. Dementia Research Center at University College London worried that analyzing tissue lysates loses valuable information about the contributions of specific cells. A recent study, for example, revealed a specific subset of microglia that surround and engulf plaques (Jun 2017 news).
Seyfried and others agreed that single-cell analysis is the way of the future. The challenge lies in the teeny amount of material in each cell. “Single-cell proteomics is advancing fast, but it’s not ready for high-throughput [analysis],” Seyfried said. Approaches to labeling proteins for capture and detection are improving, as are the mathematical tools for analysis, though Golde cautioned that every additional manipulation adds cost and contributes noise to the data.
Konrad Beyreuther from Heidelberg University was concerned that the analysis misses insoluble proteins, and Seyfried agreed this should be addressed. “The insoluble proteome likely holds a lot of clues,” said Golde, noting that changes in protein aggregation would be of interest.
Seyfried and Levey are open to sharing their data. “We want to get it out to the public as soon as possible,” said Seyfried. He plans to submit to Scientific Data Nature, an open collection of peer-reviewed data sets. Seyfried said he is also hoping Sage Bionetworks in Seattle will help link his proteomics data sets with transcriptomics data sets. Reiman and Golde were enthusiastic about such resources. “I think these rich repositories will be mined for years to come,” said Golde. “It will allow us to work faster and prevent duplicative efforts.”—Marina Chicurel
No Available Comments
Researchers convened last month in Heidelberg, Germany, could have been forgiven for thinking they had been teleported back to AD research of the 1990s. Classic, good old biochemistry captured the limelight at Mechanisms in Neurodegeneration, an EMBO/EMBL symposium. Researchers shared new data on the properties and processing of amyloid precursor protein and its fragments. They evoked new insights into γ-secretase function to explain the different potencies of various presenilin mutations and to envision γ-secretase inhibitors of the future that might spare everything bar the amyloidogenic C-terminal fragments of APP. A new candidate for the long-elusive function of APP aired when Bart De Strooper, who co-organized the meeting with Karin Dumstrei, Todd Golde, and Christian Haass, claimed that APP modulates GABA neurotransmitter receptors. Evidence that an astrocytic protease might dispatch up to half the Aβ normally produced in the brain generated a buzz.
Do Astrocytes Degrade Aβ?
Researchers led by Taisuke Tomita, University of Tokyo, homed in on the latter topic when studying Aβ degradation in cell culture. In Heidelberg, Tomita described how adding Aβ-laden conditioned medium from 7PA2 cells to neuron-astrocyte co-cultures led to reduced Aβ levels after a dozen days in vitro. During the same period, astrocytes grew in number, suggesting they may have a role in clearing the peptide. Looking at individual cell types, Tomita found that neither MG6 nor BV2 microglial lines were able to degrade Aβ, but the CCF-STTG1 astrocytoma cell line did. Tomita blocked this degradation with the inhibitor TPCK, hinting a chymotrypsin-like protease was responsible. But which one? Backed by a slew of in vitro and in vivo data that researchers at the meeting called convincing and complete, Tomita made the case that it was kallikrein 7, aka KLK7.
Previously, Patrick Daugherty and colleagues at University of California Santa Barbara had reported that KLK7, a serine protease with a unique chymotrypsin-like activity, cleaved the amyloidogenic core of Aβ (see Shropshire et al., 2013). Those experiments were based on phage display, and heretofore, no one had linked KLK7 to AD pathology, said Tomita.
Using MAB2624, a monoclonal antibody to the protease, Tomita found that astrocytes, but not microglia, expressed KLK7. Using postmortem brain samples from Japanese people aged 60-90, he found there was less of the protein in AD than in control brains. Turning to mice, Tomita asked if the protease affected amyloidosis. He crossed KLK7 knockouts with APP knock-ins created by Takaomi Saido at RIKEN in Wako, Japan (Saito et al., 2014). The crosses had more soluble Aβ in the brain than controls, and 3.5 times as many Aβ plaques. Phosphorylated tau, as determined by AT180, ticked up 20 percent. Interestingly, the KLK7-negative mice had more β-secretase around plaques, and astrocytes were more active than in controls, as well.
Do astrocytes upregulate KLK7 in response to amyloidosis? Tomita found that Aβ boosted expression of KLK7 in astrocyte cultures, suggesting the protease responds homeostatically to local Aβ levels. He looked for other signals that might do the same. Lipopolysaccharide had no effect, suggesting that general inflammatory signals hold no sway over KLK7, but glutamate, long recognized as a potential excitotoxin in neurodegenerative conditions, suppressed the protease, said Tomita. So too did NMDA, the glutamate receptor agonist. In fact, Tomita reported that the NMDA antagonist memantine, which is approved for treating mild to moderate AD, increased astrocyte production of KLK7 eightfold.
Researchers at the meeting called Tomita’s data impressive. Some wanted to know if KLK7 degraded just Aβ monomers. Tomita said so far he has only tested Aβ from 7PA2 cells developed by Dominic Walsh, which release monomers and oligomers (Walsh et al., 2002). Daugherty had also reported that treating oligomers with KLK7 prevented their toxicity. Others wanted to know if knocking out KLK7 protected the APP mice from neurodegeneration. Tomita said that work is ongoing, but the animals are too young to assess. Konrad Beyreuther, University of Heidelberg, asked why memantine does not reduce amyloid in AD patients if it so strongly increases levels of the protease. Tomita said he thinks the drug is given too late to see an effect. He suggested that early in AD astrocytes become fatigued and stop producing the protease.
Shifting the focus to Aβ production, Lucía Chávez-Gutiérrez from VIB/KU Leuven, and Harald Steiner from Ludwig-Maximilians-University, Munich, shared new data on γ-secretase function. Chávez-Gutiérrez offered an explanation for why so many different familial AD mutations increase the production of Aβ42 over Aβ40. In APP the most aggressive mutations are in the transmembrane domain that interacts with presenilin, but others are scattered throughout the protein, suggesting the global structure helps dictate pathogenicity. Likewise, in presenilin 1, FAD mutations are scattered throughout the protein, meaning most of them cannot affect the active site of the enzyme directly. Instead, Chávez-Gutiérrez proposed that they weaken the stability of the enzyme-substrate complex. To test this, she used a simple trick. She raised the temperature of the enzyme assay on a hunch that mutant enzyme-substrate complexes would more readily dissociate as the temperature rose than wild-type complexes would. This would limit processivity—that is, γ-secretase’s ability to chop amino acids off the C-terminal end of the APP transmembrane domain. Michael Wolfe at Brigham and Women’s Hospital, Boston, had previously reported that γ-secretase made more of the longer Aβ42/Aβ43 peptides as the temperature heated up, but hadn’t looked at how FAD mutations affected this (Quintero-Monzon et al., 2011).
Chávez-Gutiérrez found that on turning up the heat to around 45 degrees Celsius, ε cleavage was hardly affected. But processivity fell, resulting in production of longer Aβ peptides, including Aβ43. She saw the same pattern for presenilin mutants; however, the shift toward longer peptides occurred at lower temperatures. Instability of the enzyme-substrate complex and longer Aβ peptide production correlated with a mutation’s pathogenicity, as judged by its expected age at disease onset. De Strooper, now at the U.K. Dementia Research Institute at University College London, considers the findings important. “This shows us that destabilization of presenilin lies at the heart of the pathogenesis,” he said. “Finally, we have a mechanism [for presenilin mutations] that translates into a clinical outcome.” Haass, from the German Center for Neurodegenerative Diseases in Munich, agreed. “The data are super nice and show how FAD mutations have subtle effects on protein structure,” he told Alzforum. He noted that it fits well with Steiner’s work.
For his part, Steiner remained unconvinced that weakened enzyme-substrate stability fully explains pathogenicity. “There is definitely a correlation that fits and is consistent, but whether it is generally the case, I’m not so sure,” he told Alzforum. He noted, for example, that there are also mutations within the N-terminal and mid-Aβ domain that increase the aggregation propensity of the peptide. He also found it puzzling that temperature didn’t affect ε cleavage as much as processivity.
Chávez-Gutiérrez thinks that’s because the mutations do not alter the affinity of APP substrates for γ-secretase but do promote dissociation, thereby reducing processivity. Others had shown that presenilin mutations enhance dissociation, aka off rate, of Aβ42 (Okochi et al., 2013). Chávez-Gutiérrez said something must be happening after the formation of the enzyme-substrate complex and before cleavage. “A potential scenario is that mutations, and temperature, have an impact on the stabilization of the transition state, which involves structural rearrangements on both enzyme and substrate. In some cases, those structural changes could lead to dissociation instead of catalysis,” she said.
Steiner thought this was probably the case. This could explain how certain mutants, such as PS1 M139V, generate longer Aβ peptides than wild-type presenilin but have normal endopeptidase (ε cleavage) activity (Chávez-Gutiérrez et al., 2012). Wolfe had also reported that activity, i.e., ε cleavage, and processivity can be dissociated by changing not only temperature but pH and salt concentration.

Three Steps to Aβ. APP substrates bind the γ-secretase complex in step-wise fashion, latching onto nicastrin/pen-2 first (left), before sidling onto an exosite on presenilin (middle), and then finally finding the active site (right). [Courtesy of Harald Steiner.]
In fact, substrates initially bind to γ-secretase in an exquisite choreography involving three of the complex’s subunits, as previously detailed by researchers in Steiner’s lab. Incorporating photo-labile amino acids into various positions in APP, they mapped its binding to γ-secretase. The C99 APP fragment generated by β-secretases binds first to exosites distinct from the catalytic site (Fukumori and Steiner, 2016). The substrate latches on first to pen-2/nicastrin, then to the N-terminal of presenilin before finally nestling in at the active site (see image above).
Does C83, the product of α-secretase cleavage of APP, bind to γ-secretase exosites in the same way, or even bypass them, given that its ectodomain is so short? Again, Steiner and colleagues used photo-affinity tags to map C83 binding to γ-secretase. While they found that it, too, bound to exosites before moving to the active site, the amino acids involved in binding were not the same. “We might be able to exploit this difference to develop a therapeutic that selectively blocks binding of C99 to γ-secretase,” said Steiner. He showed how this might work in principle. He could block binding and subsequent cleavage of C99 by mutating residues in the ectodomain that are not found in the shorter C83. “Conceptually, by targeting exosite-binding of C99, we could get a drug that spares C83 and Notch,” he suggested.
A Receptor for sAPP?
Turning to the other end of APP, De Strooper reported that Heather Rice, a postdoc in his lab, and Joris De Wit at KU Leuven, may have finally fingered a receptor for sAPP, the soluble ectodomain shed by β-secretase. Researchers have long hypothesized that this domain might trigger some type of cell signaling but a strong receptor candidate has not emerged. Given that APP concentrates in synapses, De Strooper and colleagues used a proteomic approach to screen for sAPP binding partners there and nabbed GABAb1 receptors as having high affinity for the ectodomain.
De Strooper explained how they narrowed down the interaction to GABAb1a, one of two isoforms of the receptor. Only GABAb1a contains so-called sushi domains, and the first of these turns out to be required for sAPP binding, De Strooper said. Sushi domains appear to traffic GABAb receptors to axons (Biermann et al., 2010). A 17 amino acid motif on the extension domain of sAPP appears to recognize the sushi domain.
Does sAPPb regulate the GABA receptor? Adding the fragment to hippocampal neurons reduced mini excitatory postsynaptic potentials. It had the same effect on CA3-CA1 circuits in hippocampal slices, De Strooper said. The findings suggest that sAPP has a positive effect on GABAb receptors to reduce synaptic transmission downstream.
Steiner deems this an interesting development and others agreed. Haass called it “remarkable,” noting that people have been looking for an sAPP receptor for many years in what has proven to be an extremely difficult search. “I consider this a really important finding,” he said.—Tom Fagan
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.