You thought Alzheimer’s was complicated? Or Parkinson’s? Try dementia with Lewy bodies, the common disease occupying the territory between those two poles. DLB is heterogeneous and often overlooked. Yet the International Dementia with Lewy Bodies Conference held earlier this month in Fort Lauderdale, Florida, showcased a field coming into its own. For four days, four hundred researchers, patients, care partners, and other stakeholders traded new insights. Because this conference last convened about a decade ago, there was much to learn. Over the din of disease classification debates that perennially accompany spectrum diseases, the meeting reflected a field trying to build on a broadly accepted consensus diagnosis, its unifying foundation. Pharmaceutical companies are dipping in their toes, GWAS are ramping up, and prodromal biomarker cohorts are forming. Farthest ahead is Japan—the only country that has annual DLB conferences, an approved medication, and arguably the best diagnostic scan standardized across centers.
Dementia with Lewy Bodies: Is the Research Ready For Clinical Trials
Marked by degeneration of both the mind and the body, dementia with Lewy bodies (DLB) has for the past 20 years languished on the margins of medical research as an overlooked, mysterious hybrid of Alzheimer’s and Parkinson’s diseases. Largely unrecognized as a disease of its own, DLB has tended to be classified as one of its larger, eponymous cousins depending on whether a patient first sought a diagnosis for his or her symptoms from a dementia or a movement-disorder specialist—that is, if they saw a specialist at all. In fact, getting a DLB diagnosis still typically means an 18-month odyssey from doctor to doctor, and misdiagnoses along the way. However, as the International Dementia with Lewy Body Conference held December 1-4 in Fort Lauderdale, Florida, made abundantly clear, the field is shedding its relative obscurity.
In DLB, Lewy bodies (large pink sphere) form in neurons in the brain stem (top), the cortex (bottom), and other areas of the nervous system). [Courtesy of Kenji Kosaka.]
Hosted jointly by the Mayo Clinic and the Lewy Body Dementia Association, the meeting drew 400 researchers, patients, caregivers, funders and related stakeholders from across the world for a packed, 12 hour-a-day program. Attendees exchanged data in 55 talks, 117 posters, and evening symposia, while working groups of leading clinicians and scientists tried to forge consensus on how best to diagnose, manage patients, establish biomarkers, and harmonize worldwide research on DLB going forward. Patients and their care partners convened in a parallel track, but they also met with panels of clinicians and wove in and out of the scientific sessions to pepper researchers with questions.
Covering all aspects of DLB, the conference generated an overall sense that scientists have made progress in grasping this multifaceted disease. This has persuaded the pharmaceutical company Axovant to begin testing two investigational drugs specifically in DLB starting in 2016. Until recently, scientists across academia, industry and—key for drug development—at the Food and Drug Administration questioned whether DLB truly was a unique disease that needed its own indication and reimbursement code. In that regard, the psychiatric diagnostic bible, the Diagnostic and Statistical Manual of Mental Disorders, did the field a favor by adding “Mild or Major Neurocognitive Disorder with Lewy Bodies” into its disease-classification system to its current, fifth version. This gives DLB an insurance reimbursement code and helps validate its identity, said Ian McKeith of Newcastle University, U.K.
In the absence of drugs approved for DLB, practicing clinicians who recognize the disease are using various drugs indicated for AD, PD, or psychiatric and sleep conditions to manage the complex and sometimes contradictory needs of their DLB patients, said Zuzana Walker of University College, London. While allowed, pervasive off-label use creates unease among prescribing physicians, McKeith said. Clinicians need not only stronger evidence of how approved drugs perform in DLB, but also new drugs being evaluated specifically in DLB, agreed Bradley Boeve of the Mayo Clinic in Rochester, Minnesota, who co-organized the meeting. This conference series first offers a primer about DLB, and then highlights selected topics.
What, Exactly, Is DLB Again?
You might be forgiven for asking. As are AD and PD, DLB is a Lewy body disease marked primarily by pathologic aggregates of the synaptic protein α-synuclein, which are thought to spread from brain area to brain area over the course of years. However, DLB is heterogeneous in many ways. First, it is not a disease of a single neurotransmitter system, in the way Alzheimer’s is primarily seen as a cholinergic and PD as a dopaminergic disorder. Besides those two neuron types, Lewy pathology in DLB also settles in noradrenergic and serotonergic neurons, said Dennis Dickson of the Mayo Clinic in Jacksonville, Florida. In fact, the only major neuron type that seems to escape α-synuclein deposition in DLB are GABAergic ones, said Glenda Halliday of the University of New South Wales, Sydney.
In DLB, α-synuclein pathology comes in many shapes besides the signature, large Lewy body next to the nucleus (a, b). Small Lewy bodies and other aggregates in neurites (c, d) are even more abundant. [Courtesy of Dennis Dickson, Mayo Clinic.]
Second, DLB is not a focal disease. It attacks areas across the brain, from the olfactory bulb in the front to the visual cortex in the back, from the neocortex on top to the amygdala and brain stem deep within. DLB isn’t just a brain disease, either. Branches of the autonomic nervous system innervating the heart, the intestine, the bladder, and even sex organs also degenerate, slowly erasing the function of the central and peripheral nervous system.
DLB attacks many parts of the central and peripheral nervous system. [Courtesy of Bradley Boeve.]
This leaves patients and caregivers to grapple with a forever-growing list of symptoms in six different functional domains, and with frequent health crises and trips to the emergency room where medical staff are sometimes unaware of the full spectrum of DLB symptoms.
DLB’s overlapping symptoms fall into six functional domains. [Courtesy of Bradley Boeve.]
Third, DLB is heterogeneous in the sense that not every patient has the same, or all, DLB symptoms. Some suffer most from decline in the cognitive (i.e., impairment, fluctuating attention, dementia) and motor domains (i.e., the slow and stooped, shuffling walk of parkinsonism). Others are tortured by neuropsychiatric demons (visual hallucinations, delusions, depression, and anxiety), and/or suffer from urinary incontinence, constipation, and erectile dysfunction. Some decline rapidly in the face of orthostatic hypotension that starves the brain of blood and renders them prone to falls. Many DLB patients have lost their sense of smell and some see colors differently. Almost all DLB patients have sleep problems. They act out dreams at night, sometimes injuring their bed partner, are sleepless at night, and excessively drowsy during the day. “I have not met a DLB patient who does not have some sleep disorder,” said Boeve. Sleep is regulated in parts of in the brain stem and hypothalamus, regions riddled with α-synuclein pathology early on in PD and many instances of DLB.
The type of dementia in DLB differs from that of AD in that language and memory often remain relatively intact but attention, executive, and visuospatial function get worse. An engineer will lose his facility with home electronics, a planner her ability to coordinate events.
Many of these symptoms can occur in Alzheimer’s or Parkinson’s or indeed some psychiatric conditions, as well, and this overlap is one reason why many DLB cases are incorrectly diagnosed. To lay down a DLB pattern, clinicians have appointed dementia, cognitive fluctuations, visual hallucinations, parkinsonism, and REM sleep behavior disorder (RBD) as core features that distinguish DLB from other conditions. Consensus criteria published 10 years ago organize the list of symptoms into a diagnostic algorithm in an effort to capture as many different symptom combinations a given patient has at the time of diagnosis (McKeith et al., 2005). These criteria have worked reasonably well overall even though many cases are still missed, Boeve and Dickson said. In the wake of the Fort Lauderdale conference, they will be updated to include new insights, likely giving more prominence to sleep disturbances and cardiac diagnostic imaging.
DLB Research—A Thumbnail History
Research in this young field started in Japan and the United Kingdom. In Japan, Kenji Kosaka at Yokohama City University School of Medicine first connected Lewy bodies—which were thought to be the province of Parkinson’s alone—with dementia. Kosaka had been reporting in the ’70s what he then thought were isolated cases of people whose disease started with tremors and forgetfulness and progressed to dementia and parkinsonism, and who had both cortical Lewy bodies and AD pathology in their brain, (e.g., Kosaka et al., 1976).
This drawing by Kenji Kosaka details the distribution of Lewy pathology in a 65-year-old woman, his first case published in 1976. [Courtesy of Kenji Kosaka.]
Kosaka subsequently published the first European DLB cases while on a sabbatical in Munich in 1979. In 1980, back home, he proposed the term Lewy body disease along with three flavors: “brain stem” (what clinically shows up as PD), “transitional,” and “diffuse.” The latter two present clinically as DLB, and have more broadly distributed α-synuclein deposits than the former.
Two oceans away, in Newcastle, McKeith started realizing in the early 1980s that some presumed Alzheimer’s cases must have something else. In Fort Lauderdale, he recalled a 70-year-old man with an expressionless face who was moody, apathetic, and sleepy, then started seeing soldiers in a tree outside his house, walked with a stooped gait, and had trouble copying simple drawings. He deteriorated rapidly, indeed fatally, on haloperidol, and his brain proved to have been full of not only AD pathology but also Lewy bodies.
In the 1990s, Kosaka reported that DLB most commonly occurs together with Alzheimer’s plaques and tangles and more rarely as a pure α-synuclein pathology. McKeith realized that a clinical phenomenon was emerging internationally but everyone was calling it different terms, so in 1995 he invited Kosaka and other leading clinicians to gather for a first International Workshop on Dementia with Lewy Bodies in Newcastle. Numbering 170 at the time, the budding DLB community formed a consortium. They hammered out initial consensus criteria to diagnose DLB, published them together (McKeith et al., 1996), and met again three years later at the International Conference on Alzheimer’s Disease in Amsterdam to interest the broader world community of dementia researchers in DLB. At this point the DLB consortium decided that depression and sleep disorders belonged in the evolving definition of the disease, as well.
In 2003, the DLB pioneers met again and improved the diagnosis and management criteria (McKeith et al., 2005). By providing a consistent and stable classification and terminology, this so-called “Third Report” has since become the touchstone of DLB research.
In 2006, a meeting in Yokohama introduced scintigraphy imaging with the tracer MIBG. Kosaka subsequently founded a national society for DLB research, which every November convenes Japanese DLB scientists and families to drive the research agenda forward. This engaged clinicians throughout the country, who then ran clinical trials and accomplished registration of donepezil for DLB in Japan.
DLB researchers met again for a few smaller meetings, for example at the 2006 World Parkinson’s Congress in Washington, D.C. (Lippa et al., 2007), at a dedicated DLB workshop organized by Brit Mollenhauer in Kassel, Germany, in 2009, and for subsequent sessions at AD/PD and elsewhere (see May 2009 conference news; Jun 2009 conference news). Except in Japan, however, interest in DLB on the part of funders, as well as the field’s publication output, flagged for a while until there was a renewed upsurge in the past year, McKeith said.
How Common is DLB?
If only researchers knew. One problem, said McKeith, is that while the consensus diagnostic criteria have proven in the past decade to be quite specific—that is, they distinguish DLB correctly from its overlapping diseases—they are not sensitive. They miss DLB in many cases where it is actually present. That is in part because many diagnosing physicians in primary, secondary, and even some tertiary care settings are untrained in DLB and most commonly diagnose AD or PD, or a psychiatric condition, McKeith said. Epidemiology surveys thus count an unknown number of DLB as something else. Published epidemiology estimates for DLB run to about 4 percent of dementia diagnosed clinically, plus 2 percent of dementia are diagnosed with Parkinson’s disease dementia (PDD), which occurs on the same pathological spectrum. Autopsy reports flag 10 to 15 percent of lifetime dementia cases as DLB, and Kosaka told the audience in Fort Lauderdale that DLB accounts for up to 20 percent of dementia in his country. DLB is more common in men than women, and tends to become overtly symptomatic in the 70s after a long prodromal stage.
How to Detect DLB?
Beyond symptoms, physicians have some help from brain imaging in telling apart DLB and PD. For example, fitting the memory preservation in DLB, the medial temporal lobe, especially the hippocampus within it, is shrunken in AD but relatively intact in DLB. This is apparent on MRIs or even CT scans, and used in routine clinical care. In research settings, FDG PET reveals characteristically reduced metabolism in the occipital cortex of the brain, which fits with the visual impairment in DLB, as well as a curious feature called the posterior cingulate island sign. Importantly, Iodine-123 Ioflupane CIT SPECT, trade named DaTscan in Europe and the United States, visualizes loss of dopamine transporters, i.e., dopaminergic neurodegeneration, in the striatum. It is approved and widely available in Europe and Japan. In 2013, CIT SPECT also got the U.S. FDA’s blessing for differentiating tremor associated with PD from essential tremor. CIT scans well distinguish DLB from AD, though less well from related diseases on the parkinsonian side of the spectrum.
While these tools are somewhat helpful where they are available, they do not always clinch a diagnosis, said Boeve. MRI and FDG PET signals miss many cases, and clinicians need more definitive tools. While the field is eagerly awaiting a PET tracer for α-synuclein, scientists are searching for better methods or multimodal combinations of current methods. In Fort Lauderdale, researchers presented new data on both (see Part 3 and Part 4 of this series). However, the standout was the scintigraphy tracer MIBG, which in the United States is sold as AdreView but not used for DLB (see Part 2 of this series).
Fluid biomarkers for DLB are retooling in that a new set of commercial α-synuclein CSF assays are being developed at the Belgian biotech company ADx Neurosciences based on recent work by Omar El-Agnaf at Hamad Bin Khalifa University, Doha, Qatar. Proteomic or lipidomic blood-based markers are being actively pursued but remain at an earlier research stage.
Much remains to be done to improve the diagnosis of DLB. In the United States, for example, a review of a formidable clinico-pathological data set compiled by the National Alzheimer’s Coordinating Center, aka NACC, showed that only 12 to 30 percent of past DLB cases were recognized even by clinicians at the federally funded network of Alzheimer’s centers (Nelson et al., 2010). Some of these NACC data precede broad awareness of DLB as a common disease. In Fort Lauderdale, experts agreed that in 2015, diagnosis in tertiary care settings works somewhat better, and some studies to validate the 2005 diagnostic criteria suggest as much (Tiraboschi et al., 2015; Fujishishiro et al., 2008). But even today, some dead giveaways, such as cognitive fluctuations, remain easily missed in clinical assessment. In particular, primary care physicians and geriatricians, with the 10 to 15 minutes they have per patient, still need better, quicker tools to alert them that their patient might have DLB and need a detailed assessment.
To that end, James Galvin of Florida Atlantic University in Boca Raton, presented two screening tools. Called Quick Dementia Rating System (QDRS), one is a three-minute questionnaire to flag the presence of dementia and stage its severity (see Dec 2014 conference story). Called the Lewy Body Composite Score (LBCRS), the other is a three-minute questionnaire to tip off the primary care provider that Lewy body pathology, rather than AD, might be the underlying cause of the dementia. The LBCRS translates both the DLB criteria and Galvin’s experience watching his grandfather develop this disease into simple questions. In essence, the questions evoke observations his grandmother might have made caring for her husband. They ask about slow movements, excessive daytime sleepiness, acting out dreams, episodes of illogical thinking. “We are not reinventing the wheel. We just operationalized the consensus criteria in a way a caregiver could give us this information without using the clinician’s time,” Galvin said.
A caregiver fills out the questionnaires while in the waiting room, or online at home before coming in. By having this information beforehand, the health care provider can use his or her time during the visit with the patient to probe deeper, and may be more likely to order a full diagnostic workup or refer to specialty care. The screen is free for academics but requires licensing through the university for commercial providers, Galvin said.
If the Diagnosis Is so Easily Missed, Why Bother?
Getting the diagnosis exactly right is important for many reasons, researchers agree. Not least, the broad range of symptoms of DLB compared to pure AD or PD greatly complicates the physician’s ongoing work of managing a given patient’s disease. Physicians use an armamentarium of cholinesterase inhibitors, dopaminergics, glutamate antagonists, atypical antipsychotics, sedatives, antidepressants, sleep- and wake-promoting agents, and other drugs, because many of the individual symptoms of DLB are quite treatable. However, in doing so doctors and carers navigate a minefield of potential adverse effects.
For example, many people with DLB suffer from delusions and visual hallucinations that warrant treatment. The latter are particularly common and bothersome in DLB, and making them less frequent and less intense is a key goal of drug therapy, said Boeve. But DLB patients can be supremely sensitive to traditional neuroleptic drugs. Believing, erroneously, that a patient has AD, and treating him or her with an antipsychotic for delusions and hallucinations, which also occur in AD, can pitch a person with DLB into a severely rigid state, and sometimes a life-threatening condition called neuroleptic malignant syndrome can occur. In Fort Lauderdale, clinicians extensively debated the use of antipsychotic drugs for DLB, and agreed that even in the face of a pressing medical need, overall both their safety and effectiveness is questionable.
In addition, people with DLB cannot tolerate the anticholinergics often given to the elderly for urinary incontinence. Some need dopaminergic agents for their parkinsonism, but too much of those can worsen their hallucinations and autonomic symptoms.
Managing a DLB patient’s manifold symptoms is complicated, because many drugs used to treat one symptom can worsen another. [Courtesy of Bradley Boeve, Mayo Clinic.]
Sleep disturbances are more straightforward to treat, as both sleep-inducing and wakefulness medicines can be safe and effective. In Fort Lauderdale, Maria Lapid of the Mayo Clinic in Rochester reported results from a 12-week, open-label study of the wake-promoting drug armodafinil. This medication appeared to not only help patients stay awake during the day, but also lessen their visual hallucinations, agitation, and apathy. Caregivers, in turn, reported better quality of life. The Mayo researchers have been unable thus far to interest the drug maker in sponsoring a randomized controlled trial to confirm these data, Boeve said.
“Managing DLB patients is challenging. That is not a cliché. Clinically, every patient is different, and what you prioritize is different and changes over time. In the U.S. there are no FDA-approved agents, so we use everything off-label. There is little formal evidence, and no drug comes without negatives,” Boeve summed it up.
Ironically, while the acetylcholinesterase inhibitor donepezil was originally developed and licensed for Alzheimer’s, researchers in Fort Lauderdale claimed that it appears to work a little better in DLB than in AD, perhaps reflecting DLB’s significant cholinergic component. In Japan, a nationwide multicenter trial of donepezil in DLB led to its registration for DLB, the only instance worldwide of a drug licensed for DLB thus far (Mori et al., 2012; Ikeda 2013; but see also Ikeda et al., 2015). That is but a first step, however. “We really need new targets and new drugs, to treat DLB,” said Clive Ballard of King’s College, London.
How to Get More Therapy Trials Up and Running in DLB?
To plan for more treatment studies, the field is beginning to coordinate efforts around the challenge of characterizing the prodromal phase of DLB. This would help identify and enroll early stage patients into trials, much as is being done in Alzheimer’s and Parkinson’s research.
The DLB field does not yet have the biomarker staging diagrams along a time axis of progression from prodromal to MCI to the full-blown symptomatic stages that have become a staple of AD research. But the necessary pieces are moving into place. Longitudinal MCI studies have exposed a helpful dichotomy whereby amnestic MCI tends to progress to AD and non-amnestic MCI to DLB (e.g., Ferman et al., 2013). The number of studies trying to pinpoint even earlier symptoms is rapidly increasing. Observational studies to characterize people with REM sleep behavior disorder have shown that RBD usually signals underlying α-synuclein pathology and a high risk of developing DLB or PD. Alex Iranzo of the Hospital Clinic in Barcelona, Spain, mentioned that in a study of 167 RBD patients, 91 percent developed a neurodegenerative disease within 14 years (e.g., Iranzo et al., 2014).
Other studies suggest that combinations of pre-clinical features can be found to predict which α-synuclein diseases lie in a given person’s future. Daphné Génier Marchand of McGill University, Montreal, reported that in her center’s prospective cohort of 76 people with RBD followed for 10 years, cognitive tests of executive and visuospatial function versus memory can distinguish for the purpose of clinical trial enrollment which disease a person is likely to develop. Geneticists are looking for predictive variants in this cohort, as well (see Part 6 of this series). David Salmon of the University of California, San Diego, presented new cognitive tests that tax visual processing functions that are specifically impaired in DLB and help distinguish this disease from AD (e.g., Landy et al., 2015; Landy et al., 2015).
At this early stage of research, scientists are—crudely and tentatively, as they are the first to admit—marking the time axis in this proposed order. They place RBD, depression, loss of smell and/or constipation into a first, prodromal stage. Then they add in subtle cognitive problem-solving and, for some people, parkinsonian changes into an MCI stage. They place frequent visual hallucinations and fluctuations well into the symptomatic phase. The problem here, again, is that one patient may progress quite differently than another.
In their effort to start planning for disease-modifying trials in DLB, clinician-researchers are beginning to organize data from prodromal DLB research into a draft-staging diagram. [Courtesy of Bradley Boeve, Mayo Clinic.]
To supply this proposed staging scheme with stronger empirical data, longitudinal clinical and biomarker cohorts are beginning to form. For example, the EU-funded European DLB study (E-DLB) is currently collecting existing data on 1,208 DLB patients from more than 20 centers in 13 countries. E-DLB has published a harmonized protocol and begun enrolling for a prospective cohort, but the latter effort is not yet fully funded, said Dag Åarsland of Karolinska Institute in Stockholm. Smaller local studies are ongoing, for example the LewyPro study in Newcastle. In the United States, DLB patients tend to be followed within the federally funded ADRC program and the associated NACC database, said Thomas Montine of the University of Washington, Seattle. A national cohort study dedicated to DLB has not been launched.
In Fort Lauderdale, leading researchers discussed how to harmonize assessments and share data to enable trans-Atlantic cohorts to coalesce and gain statistical power. They agree that this will prepare the field for trials of disease-modifying drugs. At the same time, some scientists who are dissecting the relative contributions of amyloid, tau, and α-synuclein pathology to the head full of protein deposits that plague DLB patients argued that some of these patients could be made eligible for trials of anti-amyloid or anti-α-synuclein therapies even now, before the proposed trial-ready DLB cohorts are established. —Gabrielle Strobel
McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M.
Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium.
Neurology. 2005 Dec 27;65(12):1863-72.
PubMed.
Through the Heart? Cardiology Tracer to Nail DLB Diagnosis
For all the brain-imaging research scientists in North America and Europe have been doing on dementia with Lewy bodies (DLB) over the past decade, could it be said they were a little slow to catch on to an option that plainly does the trick? At the International Dementia with Lewy Bodies conference December 1-4 in Fort Lauderdale, Florida, a little-known scintigraphy method wowed the audience. While international research slowly advanced on a panoply of MRI, CT, SPECT, PET, and even multimodal methods, Japanese researchers cleanly and methodically moved one method across the finish line. They performed clinical studies, standardized their method across centers, ran a multicenter trial. In 2012, bingo! The Japanese health authorities approved the test as a diagnostic aid for DLB, Parkinson’s dementia (PDD) and Parkinson’s disease, and national health insurance agreed to cover the procedure. The test in question is called 123I-Metaiodobenzylguanidine Myocardial (aka MIBG) scintigraphy. MIBG, the tracer, is an analog of norepinephrine. It binds to adrenergic nerve terminals of the postganglionic sympathetic nerve that innervate the heart muscle. Its uptake is characteristically reduced in LBD, PDD, and DLB—essentially reflecting the α-synuclein-induced sympathetic denervation that underlies some of the autonomic symptoms of DLB.
Normal (A, AD patient) and reduced (B, DLB patient) myocardial MIBG uptake (arrows) in 123I-MIBG myocardial scintigraphy. For this diagnostic technique, the ratio of tracer uptake in the heart (H) over a section of a chest space (mediastinum, M) is the measure used to deduce degeneration of sympathetic nerve terminals in the heart. [Courtesy of Masahito Yamada, Kanazawa University.]
Gamma-scintigraphy is widely available and routinely used in medicine, for example for bone scans to diagnose osteoporosis. The MIBG tracer has been available in both Europe and North America for years. In the United States it goes by the name of AdreView. It is indicated for the detection of tumors of the adrenal gland, but also as an adjunct to assess sympathetic innervation of the heart. Cardiologists use MIBG scintigraphy in the context of heart failure. Most neurologists do not, but in Fort Lauderdale, many said that they would like to start. James Leverenz at the Cleveland Clinic in Columbus, Ohio, echoed the stance of many colleagues at the conference when he said, “The cardiologists at my clinic use MIBG. I wish I could use it for the differential diagnosis of DLB.”
In the United States, one argument that has held back development of MIBG was a concern that it would erroneously indicate DLB in people with diabetes or other conditions common in the elderly. However, a multicenter study presented by Cristina Muscio of Fondazione Europea Ricerca Biomedica and Pietro Tiraboschi of Fondazione IRCCS Istituto Neurologico Carlo Besta in Milan showed that myocardial uptake of MIBG was normal in old people with concomitant diabetes or mild heart disease consistent with New York Heart Association classes I or II. This suggested to Tiraboschi and Muscio that the concern might be overstated. Likewise, a recent Belgian study reported that MIBG worked properly in patients in routine clinical care who were not handpicked to exclude diabetes, heart disease, and other conditions (Slaets et al., 2015).
“The argument that MIBG gives a false positive read in diabetics is wrong. MIBG provides a robust, reliable measurement,” agreed Douglas Galasko of the University of California, San Diego. To better understand whether MIBG is useful in real-life clinical settings, Tiraboschi said, additional data are needed from larger samples of subjects with diabetes and/or heart disease. In response to audience questions about confounding conditions, Masahito Yamada of Kanazawa University Graduate School of Medicine in Japan clarified that only people with severe congestive heart failure and those who take certain antidepressants should not have the procedure.
In an AD patient, MIBG retention in the heart muscle is normal (top, red color in red circle). In a DLB patient, MIBG retention in the heart is abnormally low (blue color in blue circle). [Courtesy of Pietro Tiraboschi, IRCCS Milan.]
“We have MIBG at MGH,” said Stephen Gomperts of Massachusetts General Hospital in Boston. “I’d like to look into what it takes to use it for DLB.” Kejal Kantarci of the Mayo Clinic in Rochester, Minnesota, welcomed MIBG as a complement to the imaging toolbox that specifically captures the autonomic degeneration of this multifaceted disease.
“The MIBG data presented here look stellar. Of all imaging modalities we have thus far, it looks to be the most sensitive and specific,” said Bradley Boeve, also at Mayo in Rochester. So what were the data?
First, a note on the existing literature. Over the past 15 years, researchers at different centers in Japan and Korea have published dozens of papers building a case that MIBG myocardial scintigraphy is helpful in distinguishing dementia with Lewy bodies from Alzheimer’s and other related conditions (e.g., Yoshita et al., 2001; Watanabe et al., 2001; Yoshita et al., 2006; Nakajima et al., 2008; Kobayashi et al., 2009; and many others). Research groups in Spain, Italy, and Germany weighed in, too (e.g., Estorch et al., 2006; Estorch et al., 2008; Novellino et al., 2010; Jost et al., 2010). Meta-analyses and calls for standardization started appearing (e.g., Fröhlich et al., 2010; Teglia and Cason, 2012), and before long, there was a body of evidence from well over 100 papers, almost exclusively from Asia and Europe. The year 2015 has seen 15 papers alone on MIBG in DLB. It is not a contradictory literature. Most reports confirm the clinical usefulness of MIBG for this purpose.
At the conference in Fort Lauderdale, three presentations focused on MIBG. Yamada, of Kanazawa University, picked up from numerous single-center studies that over the years have reported high sensitivity and specificity, for an overall accuracy of MIBG scintigraphy in the mid-to-high 90th percentile. To see how that held up in a broader trial, and to put MIBG on a national footing, Japanese scientists conducted a 10-center study of 133 people with a consensus clinical diagnosis of DLB or AD.
This trial followed on the heels of a previous effort to standardize the acquisition procedure across the different camera-collimators systems in place across the country. Much as was done in ADNI years ago, a traveling calibration phantom was used to work out how to harmonize the values generated across 84 Japanese institutions and convert them to a unified H/M ratio, the main output measure of MIBG scintigraphy for DLB (Nakajima et al., 2014). The H/M ratio is a standardized ratio of the tracer’s uptake in the heart versus the mediastinum, which is essentially the space in the chest that houses the lungs and other organs there. Previously, centers performed the procedures in their own way, taking the images at different times after the injection, calculating the H/M ratio based on different-shaped regions of interest, coming up with different cut-offs, etc. Standardization makes it much easier now to perform multicenter studies, said Yamada.
In the first major trial, three readers who were blinded to the clinical diagnosis classified each image as normal or abnormal. The trial also evaluated an automated calculation of the H/M ratio based on regions of interest. This calculation achieved a sensitivity of 68.9 and specificity of 89.1 to distinguish DLB from AD; the visual assessment came out at 68.9 and 87.0 percent, respectively. In a subgroup whose dementia was still mild, the numbers were 77.4 and 93.8 percent, respectively (see Yoshita et al., 2015). As expected, his multicenter accuracy is lower than in previous single-site studies, Yamada said, but still good enough for MIBG to be useful in routine clinical care. Other researchers at the conference strongly agreed. Ron Postuma of McGill University, Montreal, summed up a general sentiment when he said, “This is very impressive evidence, and good enough for use in diagnosis.”
Satoshi Orimo of Kanto Central Hospital in Tokyo tied low uptake of MIBG in the heart to degeneration of sympathetic nerve terminals. He conducted the kind of postmortem validation study that is generally seen to clinch the connection between an imaging signal and what it claims to represent. These studies require postmortem tissue from autopsy-confirmed patients who died not too long after having received the imaging method in question. More broadly across the field of neurodegeneration, these studies have been done for every FDA-approved amyloid PET tracer and are starting in tau PET tracers, as well.
Showing how this was done for MIBG myocardial scintigraphy in life, Orimo presented data from six Japanese sites, on heart muscle tissue from 23 people with Lewy body disease and two controls who had had Alzheimer’s or multiple-system atrophy (MSA). People with AD tend not to have orthostatic hypotension or cardiac sympathetic nerve degeneration; people with MSA do have the marked orthostatic hypotension of DLB, but theirs is due to degeneration in central and preganglionic neurons, not the postganglionic nerve that degenerates in DLB and is imaged by MIBG.
The scientists stained snippets of the heart’s left ventricle with antibodies against neurofilament and tyrosine hydroxylase, the catecholaminergic enzyme in peripheral sympathetic neurons, α-synuclein. They compared this immunohistochemistry with the MIBG H/M ratio. This measure is markedly reduced in DLB. Orimo reported a statistically significant correlation between the two measures. In essence, in cases where the H/M ratio had been down during life, the TH-immunoreactive nerve fibers were gone after death, confirming that MIBG most likely detects the loss of these axons in DLB. This study appeared in print last September (Takahashi et al., 2015). It follows previous studies linking the degeneration of these nerve endings to the presence of α-synuclein aggregates (Orimo et al., 2008).
“This clinical-pathological correlation is compelling,” Boeve told Alzforum.
Several studies have compared MIBG to 123-I-FP-CIT, aka DaTscan, a SPECT method to quantify degeneration of dopaminergic nerve terminals in the striatum. FP-CIT SPECT is available in Europe, North America, and Japan. In Fort Lauderdale, Muscio and Tiraboschi directly compared the diagnostic value of DaTscan to MIBG in 32 people with a clinical diagnosis of DLB and 27 people with a non-DLB dementia. All were referred to five dementia centers in Italy in 2012. This is the first prospective longitudinal comparison study in the field, in that people received an independently verified clinical diagnosis, both striatal and heart imaging, and then were re-evaluated clinically a year later to see if the initial diagnosis was correct or needed to be changed. The clinicians and the image readers were respectively blinded to imaging data and to clinical information until the end of the study. At baseline, MIBG sensitivity and specificity to DLB were 87 and 100 percent, respectively, and 87 and 78 percent for FP-CIT SPECT. At follow-up, clinicians changed their mind about two patients, reclassifying them from possible DLB to AD. This upped the sensitivity and specificity of MIBG to 93 and 100 percent, and for FP-CIT SPECT to 90 and 76 percent.
FP-CIT SPECT performed worse because it also picked up low striatal uptake in some of the AD and FTD patients, many of whom had symptoms of parkinsonism. Their MIBG uptake in the heart’s sympathetic nerve was normal. “MIBG resulted to be more specific for excluding dementias other than DLB. The feeling that MIBG scintigraphy has a great diagnostic value in correctly identifying DLB has recently led many neurologists in Italy’s Lombardy region to use this technique off-label to differentiate DLB from other types of dementia, especially AD,” Muscio told Alzforum.
MIBG scintigraphy could come in handy for differential diagnosis. At a clinical neuroimaging-pathology symposium held at the conference, Yamada presented a case of overlapping pathology of DLB and PSP in a woman who had dementia, depression, parkinsonism, and autonomic symptoms. She could have had either underlying disease. Her MIBG scan showed clear denervation of the sympathetic nerve, clinching a diagnosis of Lewy body disease. Orimo presented a case of a woman who had autonomic symptoms and could have had PD or MSA. Her FP-CIT scan had shown reduced striatal dopamine transporters, but that is the case in both diseases, Orimo said. Her MIBG scan showed denervation of the sympathetic nerve, for a PD diagnosis. MIBG is a biomarker for the presence of neuronal Lewy pathology in the heart, which occurs in PD and DLB, but not in related diseases including AD, MSA, FTD, CBD, or PSP, Yamada told Alzforum.
MIBG in Differential Diagnosis. Sympathetic cardiac nerve degeneration reduces MIBG uptake in α-synucleinopathies such as Parkinson’s, Parkinson’s dementia, dementia with Lewy bodies, and pure autonomic failure (blue). This is not the case in controls or other diseases such as multiple-system atrophy, progressive supranuclear palsy, corticobasal degeneration, vascular parkinsonism, essential tremor, Alzheimer’s, and frontotemporal dementia. [Courtesy of Satoshi Orimo, Kanto Central Hospital.]
Last August, a study by researchers at the University of Tokyo reported that the accuracy of FP-CIT SPECT and MIBG scintigraphy overall was similar in 133 patients with either AD or DLB. This direct comparison found the combination of both tests to be best. Interestingly, patients with reduced MIBG uptake had more REM sleep behavior disorder while patients with reduced FP-CIT uptake had more parkinsonism (Shimizu et al., 2016). In October 2014, researchers at Fujita Health University School of Medicine near Nagoya, Japan, reported that MIBG heart scintigraphy was superior to brain perfusion SPECT and MRI in distinguishing DLB from AD (Inui et al., 2014).
Finally, in Fort Lauderdale, scientists discussed how they could best capture prodromal DLB in their efforts to find early stage patients for future therapy trials. A recent paper by Japanese scientists had laid groundwork in reporting that cardiac MIBG uptake strongly predicted the progression from possible to probable DLB (Oda et al., 2013). Studies in Japan are offering MIBG imaging to cohorts of people who have symptoms thought to precede DLB, such as REM sleep disorder or certain psychiatric conditions. In his talk, Hiroshige Fujishiro of Nagoya University, Japan, emphasized the need to offer combined clinical and MIBG biomarker studies to middle-aged and older patients who present to psychiatrists, for example, with major depressive disorder (see also Kobayashi et al., 2015). Likewise, at the conference, John-Paul Taylor, Alan Thomas, and Paul Donaghy, all of Newcastle University, U.K., noted that MIBG imaging was a promising biomarker in prodromal DLB research (Donaghy et al., 2015).—Gabrielle Strobel
Dementia with Lewy Bodies: Sharper Image for a Formerly Blurry Disease
Dementia with Lewy bodies is a common α-synucleinopathy that blends dementia with parkinsonism and psychiatric illness. When scientists met on December 1-4 in Fort Lauderdale, Florida, for the International Dementia with Lewy Bodies Conference, the strength of Japanese data on diagnostic MIBG scintigraphy was the big surprise (see Part 2 of this series). Even so, interspersed throughout the meeting were many more incremental advances. Researchers are deploying new methods to tease apart the overlap with Alzheimer’s and Parkinson’s that has long bedeviled DLB research. Together, the insights are converging to give definition to a disease that has long struggled for recognition. This more distinct identity of DLB emerges as scientists better understand which underlying protein pathology gives rise to which of aspect of its symptoms, or to how fast DLB progresses.
Importantly, researchers are increasingly relying on imaging data and tissue from people who have been studied in life and then received a definitive neuropathological diagnosis after death. Indeed, both imaging and cerebrospinal fluid (CSF) data presented at the Fort Lauderdale conference were anchored by neuropathology. This reflects an effort to understand the heterogeneity of DLB, a disease driven by accumulation of at least three toxic proteins—α-synuclein, tau and Aβ—in different regions of the brain, at different times, in different people. A major push in the DLB field goes toward accounting for what each pathological protein contributes to the disease during life.
Three Villains.
Protein deposits of α-synuclein, tau, and Aβ plague the brains of people with DLB. [Courtesy of Tanis Ferman, Mayo Clinic.]
Dennis Dickson of the Mayo Clinic in Jacksonville, Florida, set the neuropathologic stage. He first aligned Braak’s staging of α-synuclein pathology in Parkinson’s disease—spreading from the bottom up via the vagus nerve and pons through the midbrain, basal forebrain, and eventually throughout the cortex—with categories proposed by Kenji Kosaka of Yokohama City University, Japan. (Kosaka started describing cases in 1976, and several researchers in Fort Lauderdale said that alongside the eponymous Parkinson’s and Alzheimer’s diseases, DLB might fairly have been named Kosaka’s disease).
In 1980 Kosaka had proposed three categories. The first, a restricted brainstem type, corresponds to Braak stages 1 to 3; the second, a transitional type with pathology in the basal forebrain, corresponds to Braak stage 4; the third, a diffuse type with Lewy bodies in the cortex, corresponds to Braak stages 5 and 6. Do the patients who walk into the clinician’s door fit these pathological bins, Dickson asked? Not so neatly.
In DLB, neurons in the amygdala contain both aggregated tau (brown) and α-synuclein Lewy bodies (blue). Sometimes both co-localize in the same neuron (higher magnification at bottom). [Courtesy of Dennis Dickson, Mayo Clinic.]
First off, the most common Lewy body disease is Alzheimer’s. Fully a fifth of AD cases have ample α-synuclein pathology, Dickson said. They have it in the amygdala—just as in Braak stage 4 or Kosaka transitional Lewy body disease. Their α-synuclein pathology there occurs side by side with Alzheimer’s tau pathology. “The amygdala is uniquely vulnerable to both processes,” Dickson said, while showing images of α-synuclein and tau filaments mingling even in the same neurons in the amygdala. Secondly, the olfactory bulb is also highly prone to this α-synuclein/tau double whammy, often in the same people who have it in the amygdala. The brainstem, which is emblematic of Parkinson’s, tends to be unaffected in these olfactory/amygdala cases of DLB. The amygdala and olfactory bulb are part of the brain’s limbic system.
In essence, Dickson said, Braak’s bottom-up PD staging system works largely as advertised for PD, but not for DLB. In that disease, many cases have most α-synuclein pathology in the limbic system, and it spreads from there up into both the association and primary visual cortex, and down to the autonomic system.
The distribution of α-synuclein pathology in Parkinson’s differs from that of DLB. In DLB, the limbic system predominates and appears to be affected early. [Courtesy of Dennis Dickson, Mayo Clinic.]
How do DLB cases shake out when amyloid pathology is added in? Seeing the patterns of three age-related proteopathies gets even more complicated. Dickson attempted to do this by plotting both tau pathology as per Braak stages and amyloid pathology as per Dietmar Thal phases against α-synuclein pathology as per Kosaka.
He did this with a Mayo Clinic series of 808 people who had had either AD, DLB, PDD, or PD. This created a complicated matrix of the bewildering variability of these diseases. Still, some themes emerged:
In people who present clinically as DLB, pure Lewy body disease without AD pathology is rare. Nine out of 10 also have tangles and plaques, and six in 10 have a high burden of plaques.
Among people diagnosed with PDD, i.e., who develop parkinsonism and later also dementia, one in five have a pure α-synuclein disease. The rest have both synuclein and AD pathology, and many have a heavy amyloid burden. This disease is extremely variable, Dickson said.
Among people who present with Parkinson’s, four in 10 have no AD pathology; the remaining six have mostly a low burden of it.
This means that Lewy pathology in the cerebral cortex, particularly its limbic region, is common in DLB but minimal in Parkinson’s. In DLB, the olfactory bulb and amygdala are more prone to α-synuclein pathology than in PD. Because Lewy body disorders afflict old people, they frequently occur together with Alzheimer’s pathology, Dickson said. When their AD pathology is severe, the clinical balance of their symptoms tips toward AD. “The neuropathological diagnosis of DLB is a probability statement that takes into account the severity of both Lewy and Alzheimer pathology,” Dickson said.
You, Too, Tau?
Several speakers reported converging data on the question of what the accompanying tau tangles might be doing to a person who is living with DLB. In short, tau seems to speed up their decline. Tanis Ferman, a neuropsychologist at Mayo in Jacksonville who follows patients over time, wondered why some people with DLB decline gradually while others drop off precipitously. Analyzing neuropathology against clinical data of people who had died with DLB, Ferman first noticed that the clinical picture of DLB arose both in patients with transitional LBD, i.e., Kosaka’s more restricted distribution of α-synuclein, and with diffuse LBD, where Lewy bodies are spread throughout the cortex. In other words, cortical Lewy bodies were not necessary to make people symptomatic. That said, people with both diffuse LBD and with the most tau changed the most over time. “People with the greatest tau and synuclein burden overall declined fastest,” Ferman said.
How much pathology a person has determines how fast their disease gets worse. [Courtesy of Tanis Ferman, Mayo Clinic.]
In a second, larger sample, Ferman split transitional and diffuse cases into high and low tau burden. This unmasked more differences. People with a lot of tauopathy in addition to their synucleinopathy not only progressed fastest, they also had worse memory loss and were likelier to carry ApoE4 and be female than the DLB patients with a low tau burden. Also, they were less likely to have REM sleep behavior disorder (RBD). In essence, Ferman saw facets of DLB’s AD component via the tau pathology. (Amyloid plaques were tightly correlated with tangles in this study, hence the “high tau” cases also had high Aβ.) The polar ends of the spectrum support this, as AD is more common in women and ApoE4 carriers, and marked by memory decline, whereas PD is more common in men, with more REM sleep disorder but intact memory, and lower frequency of ApoE4. The fastest decline, said Ferman, results from having a high burden of diffuse α-synuclein deposits paired with a high burden of AD pathology.
In this sample, people with both diffuse Lewy body disease—the most severe Kosaka category of synucleinopathy—and lots of tangles declined most rapidly on a measure of cognition (red). [Courtesy of Tanis Ferman, Mayo Clinic.]
Can multimodal imaging help in the face of this complex underlying burden of mixed proteopathy? In the absence of MIBG cardiac scintigraphy, a single imaging marker is not enough to distinguish the AD and DLB syndromes clearly from each other. Researchers hope that seeing several aspects of the disease at once will make things clearer. Kejal Kantarci of the Mayo clinic in Rochester, Minnesota, is analyzing a host of imaging findings from a cohort of DLB patients who have since been autopsied. In Fort Lauderdale, Kantarci reported that, at least in her hands, multiple markers can almost completely separate the two syndromes.
Kantarci also reported that those DLB patients who turned out to have had abundant concomitant AD pathology are the ones whose medial temporal lobe (MTL) shrank in life. Very little MTL atrophy registers in group studies of DLB, but it does show up in individuals who have lots of plaques and tangles. “The higher the Braak stage, the more hippocampal loss,” Kantarci said. Hippocampal atrophy in AD is widely thought to reflect tangle pathology.
In this study of 21 patients, each with DLB or AD, three imaging methods combined distinguished the two better than each individual scan. Except for one DLB patient, all AD and DLB patients are separated using multimodality imaging measures.
With that shrinkage came shorter survival. Jonathan Graff-Radford, also at Mayo Rochester, reported that in a cohort of 167 DLB patients, those with the smallest hippocampal volume at baseline died soonest after the disease was diagnosed. Graff-Radford’s model calculated a difference in predicted survival of almost four years between the DLB patients who had a shrinking hippocampus and carried ApoE4, a genetic indicator of Aβ pathology, and those DLB patients who had a relatively larger hippocampus and no ApoE4 allele. The former could be seen to have more AD admixed into their DLB, the latter less.
Yet another hint that AD pathology worsens DLB came from multimodal imaging by Stephen Gomperts of Massachusetts General Hospital, Boston. Gomperts confirmed that AD pathology has real repercussions for progression when it co-exists with Lewy pathology. He follows patients with the overlapping synucleinopathies DLB, PDD, and PD who are getting both PiB amyloid and T807/AV1451 tau PET scans. Thus far, he sees amyloid positivity in most people with DLB. Much like Ferman saw based on neuropathology, Gomperts sees that when a person’s PiB uptake, i.e., their amyloid burden, is high, then that person’s cognition will decline faster on the CDR-sb and the MMSE. Likewise, in the people with Parkinson’s who have a positive amyloid scan, high uptake predicts faster decline from normal cognition to MCI, and from MCI to PD dementia. In contrast, amyloid did not change how fast parkinsonian motor symptoms declined, Gomperts said.
Gomperts also described an early experience with tau PET, in a cohort of 29 controls, seven people with DLB, eight with PD-MCI, and nine with cognitively normal PD. Gomperts sees the most T807 retention in people with DLB. Their signal in the inferior temporal gyrus can approach levels published for AD (Johnson et al., 2015). Gomperts noted that this signal is highly variable in the DLB patients he has scanned thus far. In contrast, he saw a weaker tau signal in the inferior temporal gyrus of PDD, PD, and controls.
Those DLB patients who are the most cognitively impaired on CDR-sb and MMSE have the most T807 retention. When Gomperts added a cortical thickness measure from MRI scans into this model, he saw a three-way interaction. In essence, as T807 retention went up and the cortex thinned out, people were more cognitively impaired.
This data was well received, though Kantarci noted that in her hands, the T807 signal imperfectly matches postmortem pathology in terms of both location and amount. She believes T807 may ignore certain kinds of tau pathology in DLB. With regard to non-specific binding of T807, Gomperts pointed out that a recently published validation study (Marquie et al., 2015) showed that T807 can also bind neuromelanin in the substantia nigra as well as certain blood products; however, this is based on putting T807 directly onto brain tissue slices, not yet on a neuropathological confirmation of people who had had scans in life.
Beyond clinico-pathological and imaging work, fluid biomarkers now also confirm that AD pathology speeds up DLB. CSF biomarkers can track DLB from the beginning, while neuropathology only gets one look at the brain in the aftermath of disease. Unlike PET, CSF tests can measure multiple proteins at the same time, and they cost less than imaging, making them well-suited for repeat assessment in large cohort studies. In Fort Lauderdale, Evelien Lemstra of VU Amsterdam reported the first results from an ongoing study of one of the largest cohorts of DLB patients in Europe. Before she showed her data, Lemstra tried to soften the opposition to lumbar puncture that is ingrained at many sites in North America by saying, “We just finished CSF collection in 1,000 patients and see a very low rate of headache or other side effects.”
CSF Aβ and tau studies in AD are well-established (see the Alzbiomarkers database for meta-analyses). As for DLB, Lemstra reported that among 111 patients with clinically probable DLB, 69 had the CSF AD profile of a low tau/Aβ42 ratio (see Duits et al., 2014). They were more likely to carry an ApoE4 allele and less likely to have a positive DaTscan than the 42 DLB patients who did not have the CSF AD signature, Lemstra said.
The DLB patients with AD CSF markers had more severe hallucinations and delusions. They performed worse on tests of memory, but better on tests of executive or visuospatial function, attention, or language. Lemstra has insufficient follow-up data thus far to see if cognition in the two groups declines at different speeds, but she does see a trend that DLB patients with AD CSF markers get placed in nursing homes faster and die sooner. “The AD pathology in DLB is clinically relevant, and can be detected antemortem with CSF,” Lemstra told the audience. This cohort is part of the Europe-wide E-DLB project (see Part 6 of this series).
Given this convergence of evidence about the presence of AD pathology in DLB, and its contribution to the real-life experience of this disease, researchers in Fort Lauderdale called for more data sharing to confirm the importance of these insights in larger samples across the field. Others said the growing evidence already argues for evaluating anti-amyloid and anti-tau drugs in people with DLB who are confirmed at enrollment to have concomitant AD pathology.—Gabrielle Strobel
Duits FH, Teunissen CE, Bouwman FH, Visser PJ, Mattsson N, Zetterberg H, Blennow K, Hansson O, Minthon L, Andreasen N, Marcusson J, Wallin A, Rikkert MO, Tsolaki M, Parnetti L, Herukka SK, Hampel H, De Leon MJ, Schröder J, Aarsland D, Blankenstein MA, Scheltens P, van der Flier WM.
The cerebrospinal fluid "Alzheimer profile": easily said, but what does it mean?.
Alzheimers Dement. 2014 Nov;10(6):713-723.e2. Epub 2014 Apr 8
PubMed.
At the International Dementia with Lewy Body Conference, held December 1-4 in Fort Lauderdale, Florida, brain imaging emerged as a promising adjunct to figuring out exactly what is going on in a patient’s brain during life. Given that the clinical diagnosis often misses DLB (see Part 1 of this series), and the pathological confirmation comes too late to help the patient, researchers are exploring imaging techniques to bridge this gap. This has created an increasingly varied research field.
MRI has been around the longest, but appears to be the least powerful in DLB. Showing MRIs from autopsy-confirmed cases, John O’Brien of Newcastle University, U.K., confirmed the general finding that the hippocampus in DLB does not shrink much. This finding has become part of the consensus diagnostic criteria. Because it is visible even on CT scans, which are widely available in routine clinical care, this finding is often used to support a diagnosis. And yet, it is not true in all cases, for example those with abundant tau pathology in the hippocampus (see Part 3 of this series). In addition, O’Brien noted more recent research findings that some cases of clinical DLB do have measurable atrophy elsewhere in the brain. This takes the form of subtle thinning in the insula area of the cortex, as well as in basal ganglia and other subcortical regions. Serial imaging in research studies suggests that this ongoing atrophy is related to concurrent Alzheimer’s pathology, O’Brien said.
Beyond structural MRI, functional imaging of the brain has generated a handful of measures—from FDG PET for glucose metabolism and rCBF PET or SPECT for blood flow to diffusion tensor imaging—that offer some consistent signals in DLB. Alas, although FDG PET is a robust technique for differential diagnosis of dementia, it is less powerful in early stages of disease, said Nicolaas Bohnen of the University of Michigan in Ann Arbor. By comparison, dopamine transporter imaging in the substantia nigra, done by 123-Iodine ioflupane SPECT (aka FP-CIT or DaTscan) is highly sensitive, i.e., it does not miss cases as easily. A DaTscan greatly improves the diagnosis of DLB, though by itself it is also not always specific; dopaminergic nerve terminals sometimes degenerate in other diseases that can masquerade as DLB, Bohnen said. Thomas Beach of Banner Sun Health Research Institute in Sun City, Arizona, agreed, “An unknown fraction of people with positive DaTscans have PSP, CBD, or MSA rather than PD or DLB. We see this at our center and elsewhere.”
Better Than the Clinician?
When a patient with early stage dementia has an abnormal FP-CIT SPECT, multimodal imaging would diagnose dementia with Lewy bodies. If his or her amyloid PET scan is abnormal but the FP-CIT is normal, it would be Alzheimer’s. (When both scans are normal, the diagnosis is frontotemporal dementia, though people with PSP are an exception because their parkinsonian tauopathy renders the FP-CIT scan abnormal, too.)
[Courtesy of Kirk Frey, University of Michigan.]
Amyloid PET, which picks up the plaques that accompany many cases of DLB (see Part 3 of this series), has further stocked up the neurologist’s toolbox. Several groups at the conference reported positive amyloid scans in a majority of DLB patients. Their amyloid burden tends to be in between that of controls and people with AD, similar to the burden seen in MCI. This jibes with the neuropathological finding of variable amounts of amyloid plaques accompanying Lewy pathology in DLB.
But it is the combination of the two that, at least in specialized research settings, outperforms even astute clinicians. For example, Bohnen described an ongoing study at UMichigan of 75 patients with diagnoses of mild dementia, agreed upon by a consensus of clinicians. Nuclear medicine neurologists made their own call based on PET scans for both dopamine terminals in striatum, and amyloid. In 2011, this study’s first paper reported that the neuroimaging diagnosis disagreed with the clinical one in 17 percent of AD cases, 29 percent of DLB, and a whopping 64 percent of FTD cases. Who was right? In Fort Lauderdale, Bohnen followed up with the news that 36 of the 75 patients had since come to autopsy, and neuropathology had validated the neuroimaging diagnosis in 33 of them (Burke et al., 2011; Albin et al., 2015). “The imaging-pathological correlation is much more accurate than the clinical-pathological correlation,” Bohnen said.
As more imaging tests come online, clinicians will be able to serve their patients better by incorporating scans into their diagnoses. Then they will be able to focus their clinical expertise on managing the manifold symptomatic challenges of DLB patients, who cannot tolerate some of the medications that might be used if the patient had AD, scientists agreed. At this point, dual scans are not approved by most insurance, though Bohnen told Alzforum that at the Veteran’s Affairs hospital where he works, both DAT and amyloid PET are done quite frequently and can be very helpful in selected patients.
Looking farther out to the research horizon, Bohnen mentioned two new imaging tracers worth watching. One is 18F-FEOBV, a PET tracer for vesicular acetylcholine transporters. In DLB, FEOBV PET generates a big signal reduction in the cortex, hippocampus, caudate nuclei, and the thalamus. “FEOBV shows marked cholinergic losses not only in cognitive but also motor-control regions in DLB, as previously shown by neuropathology,” Bohnen said (Petrou et al., 2014). At the conference, Dennis Dickson of the Mayo Clinic in Jacksonville, Florida, reminded the audience that DLB causes degeneration in many transmitter systems across the nervous system.
The second emerging method may open a window to prodromal DLB, Bohnen said. Neuropathologists believe that some cases of Lewy body pathology reach the brain by way of the enteric nervous system, possibly as a consequence of exposure to toxins in food. Some scientists believe that α-synuclein pathology in intestinal neurons can damage cholinergic terminals there and lead to symptoms such as constipation, which is common in PD and DLB years before the better-known movement and cognitive symptoms. Scientists led by Per Borghammer at Aarhus University Hospital, Denmark, have begun to visualize this process.
Like MIBG scintigraphy of the heart, a diagnostic imaging test that picks up DLB in the peripheral nervous system (see Part 2 of this series), the new Danish approach also captures peripheral neurodegeneration via reduction of an affected neurotransmitter, in this case acetylcholine. But unlike MIBG, the Danish approach uses a radiolabeled version of the therapeutic drug donepezil as a PET tracer. Donepezil binds acetylcholinesterase in intestinal nerves and is, after all, highly active in this part of the body, as legions of nauseous donepezil-treated AD and DLB patients can attest. The scientists recently described how injected 11C-donepezil flows and binds throughout the human body in healthy controls, as well as in 12 people with Parkinson’s disease. In them, 35 percent less of this tracer bound in the small intestine than in controls (Gjerloff et al., 2014; Gjerloff et al., 2015). “This is a very early, prominent signal. Imaging neurotransmitter changes or α-synuclein deposits in autonomous organs may become a game-changer in the field,” Bohnen said.—Gabrielle Strobel
Lewy Pathology in DLB Spreads Fast, Maybe From the Nose
At the International Dementia With Lewy Body Conference, held December 1-4 in Fort Lauderdale, Florida, scientists suggested that concurrent amyloid pathology in DLB explains some of the overlap between DLB and Alzheimer’s disease (see Part 3 of this series). But they also reported indications that DLB is very much its own disease, and these came from DLB’s defining component, the α-synuclein pathology itself.
Glenda Halliday of University of New South Wales, Sydney, told the audience that while the knowledge base on Lewy pathology in aging humans comes largely from Parkinson’s, research on DLB is catching up and has found distinctive features in the appearance, timing, and spread of Lewy pathology. A typical Lewy body takes about a decade to grow and condense into its classic, structured form. In Parkinson’s disease, most Lewy bodies look that way. That is especially true of the older ones in the brainstem, which have had more time to mature than the more recent ones that arose later during the pathology’s gradual spread up to cortical regions. In contrast, Lewy bodies in DLB tend to look younger and less structured, Halliday said. They are also far more prevalent all over the cortex. They sweep through the brain faster, fueled perhaps by the co-occurrence of other pathologies. While people with PD survive on average 13 years after their diagnosis, DLB patients die well within a decade, some even as early as six months, after disease is apparent.
Two cortical neurons each contain a diffuse Lewy body as typically seen in DLB (left, x400). Mature Lewy body in a brainstem pigmented neuron, as typically seen in PD (right, x1000). This Lewy body is ringed by a halo. Both are stained for α-synuclein. [Courtesy of Glenda Halliday.]
“The tempo of DLB is faster, and no matter when a patient dies, even if it’s soon after their diagnosis, they tend to have Lewy pathology everywhere,” Halliday said.
In a given person, the amount of one pathology relates to the amount of another; in other words, people who have lots of Lewy bodies tend to have lots of cortical amyloid deposits, more abnormal neurites and cell loss in the hippocampus, more striatal amyloid deposits, and more atrophy. “All these measures correlate to the degree of Lewy pathology in DLB,” Halliday said.
It is not only the amount that is different, but apparently also the route the toxic proteins take. In DLB, the nose is increasingly attracting the attention of researchers as one possible explanation. Could it be that, at least in some DLB patients, α-synuclein deposition starts in the nose and spreads from there to other parts of the brain? The olfactory epithelium, which feeds into the olfactory bulb, is only a short distance away from the amydgala. The amygdala is a main hub of Lewy pathology in DLB, which in turn relays to many of the other affected areas. Thomas Beach of Banner Sun Health Research Institute in Sun City, Arizona, has long argued that a large proportion of DLB cases do not follow Braak’s brainstem-to-cortex progression of Lewy pathology. Beach and other researchers have implicated the olfactory mucosa as a possible entry zone instead (Beach et al., 2009; Funabe et al., 2012). From the nose, Lewy bodies could easily access the amygdala and then the brainstem, Beach said in Fort Lauderdale.
Tracing the route by which pathology marches through the brain invariably invokes the hypothesis of prion-like spread, and indeed this mechanistic discussion came up at the DLB conference, as well. Patrik Brundin of the Van Andel Research Institute in Grand Rapids, Michigan, drew much notice when he showed early data of a new animal model for a progressive spread of pathology starting in the nose. This model, a wild-type mouse, might be able to recapitulate a DLB- or PD-like disease.
Nolwen Rey in Brundin’s lab used sonicated α-synuclein fibrils in a collaboration with Kelvin Luk from Virginia Lee’s group at the University of Pennsylvania, Philadelphia, and injected them into the olfactory bulb of wild-type mice. Then she waited. Would they be taken up by neurons in the olfactory bulb? Would they recruit endogenous α-synuclein, seed aggregation, and lead to release of those aggregates from the neurons’ terminals in their projection area, the piriform cortex? Would they trigger this process to repeat itself in postsynaptic neurons there?
This schematic of the mouse brain depicts primary connections (red) going from the main olfactory bulb (MOB) to the piriform cortex (PC), the frontal cortex (FC), the amygdala (Am) and the entorhinal cortex (Ect). Green areas show inclusions stained with antibodies against phosphorylated α-synuclein (micrograph inserts) one month after fibril injection; orange areas, which involve traversing one synapse, three months after injection. [Courtesy of Nolwen Rey.]
Indeed, that is precisely what Rey is beginning to see, Brundin told the audience. This is ongoing work, but Brundin showed some slides indicating how, one month after the injection, α-synuclein aggregates appeared in immediate olfactory bulb projection areas. By three months, more aggregates showed up in the brain nuclei one synapse away (see image). By six months, aggregates appeared in additional areas two synapses removed from the olfactory bulb. A year after the injection, Rey saw α-synuclein deposits in about 60 brain areas and subregions that are anatomically connected, directly or indirectly, to the olfactory bulb. They are thioflavin-positive aggregates that share common markers with Lewy bodies, Brundin said. What’s more, the mice showed evidence of a progressive inability to distinguish scents, and to remember odors they had smelled before.
This spread is similar from mouse to mouse, and predictable in its timing. “There is not a lot of variability in this model,” Brundin told Alzforum, “It is very staged, and goes from the olfactory bulb to the amygdala. We have seen in some cases that it even reaches the brainstem.”
Brundin’s talk came after Virginia Lee of UPenn had set the stage for a discussion of the seeding hypothesis as an underlying mechanism. Lee focused on injections into the striatum, not the olfactory bulb. She recapped her lab’s prior work, which recently continued with a paper reporting a staged spread of α-synuclein pathology and neurodegeneration triggered by preformed fibrils in rats (Paumier et al., 2015; Nov 2012 news). “Our lab has done many injection experiments by now. We find that wherever we inject α-synuclein, its spread retraces that area’s anatomical connectome in exquisite detail,” Lee said. The seeding hypothesis is gaining ground across the field but is not uncontested (Apr 2014 news).
Brundin emphasized that templated seeding and spread starting in the nose could lead to both DLB or PD. Many people with either disease report having lost their sense of smell years before other symptoms of DLB or PD became apparent.
The idea that DLB can start in the nose is provocative but remains controversial, commented Bradley Boeve of the Mayo Clinic in Rochester, Minnesota. While he called Brundin’s model “compelling,” he also noted that many DLB and PD patients develop REM sleep disorder well over a decade before core DLB symptoms crop up. This, to Boeve and others, implies that pathology in the brainstem must start early. Researchers do not know yet if α-synuclein travels from the nose to the amygdala in some people and from the enteric system through the vagus nerve to the brainstem in other people. The clinical heterogeneity of α-synucleinopathies leaves room for multiple different paths in different people. In discussion, Brundin noted that because humans swallow their nasal secretions, both the nose and the gut in theory could be assaulted by the same environmental toxins, such as inhaled pesticides or metal fumes in welding.
“All hypotheses in DLB are difficult to test in humans without a more sensitive and specific biomarker,” Boeve said. Indeed, longitudinal biomarker studies are needed to chart the paths α-synucleinopathy takes and to understand the exact differences between DLB and PD. Until then, Brundin quipped, the relationship between the two evokes the Thai idiom “Same same, but different.”—Gabrielle Strobel
Genetics of DLB: Setting Up to Fill a Mostly Empty Canvas
Besides ample new data presentations, the International Dementia with Lewy Body Conference December 1-4 in Fort Lauderdale, Florida, featured a reprise of the field's perennial debate. It is the verbal tug-of-war between movement disorder and dementia specialists about whether research on the spectrum of Lewy body disorders is well served by clinical classifications such as dementia with Lewy bodies (DLB) and Parkinson’s dementia (PDD). Perhaps a different scientific approach can inform this old parley with fresh information? Indeed, what do geneticists say about DLB as a unique entity? Thus far, they know little, but several presenters showed how the question is starting to draw attention on a larger scale.
Jose Bras of University College London, U.K., reminded the audience that late-onset Alzheimer’s and Parkinson’s were not considered genetic diseases until recently. By now some 30 genes are known to be involved in each of them, and their overall heritability is estimated at 60 and 28 percent, respectively. At the level of GWAS, the known common genetic risk factors for AD and PD do not overlap (Moskvina et al., 2013). Yet there is DLB, a clinical hybrid of the two that is, at least according to its proponents, as common as it is overlooked and underserved. DLB genetics is just beginning to pick up steam, with some 100 papers in the literature compared with 1,000 on the genetics of PD and 1,800 on that of AD.
To date, scientists have implicated four genes in DLB. They are:
APOE, which was first reported 21 years ago and subsequently confirmed (Hardy et al., 1994).
SNCA (encoding the α-synuclein protein) by way of a clinical DLB presentation in some PD families carrying a gene triplication (Singleton et al., 2003).
GBA, variants of which drive up a person’s risk of developing DLB (Tsuang et al., 2012).
These first clues come from research with single families or samples of fewer than 100 patients.
About 1,400 pathologically confirmed cases from three continents make up an ongoing GWAS of DLB. [Courtesy of Jose Bras, Rita Guerreiro, UCL.]
To inject more power into the gene hunt, Bras and collaborators in Europe, Australia, and across North America have thus far pooled 1,400 cases confirmed by neuropathology to have had DLB. This is still a far cry from the nearly 100,000 people whose samples are available for AD/PD GWAS, but it’s a first step toward a comprehensive look at the genetic signature of DLB, Bras said. This ongoing project uses the NeuroX array, a new tool geneticists including Bras designed for rapid and cost-effective genotyping in neurodegeneration research (Nalls et al., 2015).
Samples of this size support heritability estimates. These calculations put DLB’s genetic component at 31 percent, close to that of PD and half that of AD, Bras told the audience. They also enable estimates of the genetic overlap among AD, DLB, and PD—diseases already known to overlap at the clinical and the neuropathological level. Genetically speaking, DLB correlates strongly with AD, though much of that is due to ApoE. Subtracting ApoE leaves DLB equally correlated to AD and PD, Bras reported. In contrast, AD and PD in this calculation again came out to be genetically uncorrelated. This research appeared in Neurobiology of Aging online last month (see Guerreiro et al. 2015).
Last December, Bras, collaborator Rita Guerreiro of UCL, and colleagues around the world reported preliminary results from an early data freeze of 750 samples from this growing cohort (Bras et al., 2014). This initial analysis did not survey the whole genome in an unbiased manner, but rather examined 54 genomic regions previously associated with PD and AD. Extracting variants within 500 kilobases of the known top hits from AD and PD GWAS, this study reproduced APOE as the strongest signal but also showed study-wide significant signals around SNCA, the gene for α-synuclein, and SCARB2, a lysosomal gene implicated in PD.
Because the gene chip used in these GWAS only pointed to APOE without distinguishing between the ApoE2, 3, and 4 alleles, Guerreiro decided to nail down the precise nature of the link. To do that, she harked back to the early days of DNA sequencing. She isolated DNA from the DLB patient samples, digested it with a restriction enzyme, loaded the fragments onto a gel, and compared their relative sizes to deduce the APOE genotypes. This direct genotyping of 679 DLB DNA samples showed that the APOE hit in previous GWAS is indeed due to ApoE4, Guerreiro reported in Fort Lauderdale. Specifically, the frequency of the ApoE4 risk allele in the DLB cohort, at 28.1 percent, lay midway between that of the general population (13.7 percent) and that in AD (36.7 percent). Conversely, the protective ApoE2 allele in DLB, at 5.4 percent, lies midway between its frequency in the general population (8.4 percent) and people with AD (3.9 percent). ApoE4-carrying DLB patients also tended to die younger, Guerreiro reported.
As for SNCA and SCARB2, even though these genes had come up in previous PD GWAS, a deeper look at their respective genomic regions pinpointed different individual markers in both the SNCA and SCARB2 genes for DLB and for PD. In other words, even though both genes are involved in both diseases, the most significant markers appear to be in different areas of those genes in DLB and PD. “We do not know what this means functionally yet, though it may have to do with differential control of gene expression,” Bras said.
Beyond this, the researchers are carrying out a full-fledged GWAS using the newer NeuroX2 array, which is stocked with additional, lower-frequency variants to drill deeper into the genetics of DLB. Giving a status update on this ongoing work, Bras said that to date, genotyping of 1,200 pathologically proven cases has replicated the known genes and dug up additional hits he deemed too preliminary to name. In addition, ongoing exome sequencing of the entire cohort plus some familial cases thus far has generated nearly 10,000 new, high-impact variants of yet-unknown clinical significance. Some are in genes known to be involved in AD and PD, such as GBA, PSEN2, SCARB2, TREM2, GRN, TYROBP, APOE, and LRRK2, Bras showed.
In his talk, Ziv Gan-Or of McGill University in Montreal echoed the theme of the same genes showing up in overlapping diseases by way of different markers. In Fort Lauderdale, Gan-Or described an ongoing sequencing and genome-wide association study of 1,200 people with REM Sleep Behavior Disorder and as many controls. RBD is increasingly seen as a prodromal stage of DLB or PD. While this work is ongoing, Gan-Or noted that the genetics of people with this sleep disorder is emerging to be similar overall to that of people with DLB. Some of the same genes that are known from previous genetic studies of α-synucleinopathies are coming up in this RBD GWAS. In particular, several known PD- or DLB-linked mutations in GBA and SCARB2 were associated with RBD. In the future, genetics and clinical data together may be able to identify people with this sleep disorder who are likely to progress to DLB or PD within the time frame of a clinical trial (see Part 1 of this series). Data from a smaller study leading up this the current project were recently published (Gan-Or et al., 2015).
The genetics of dementia with Lewy bodies, unlike that of Alzheimer’s or Parkinson’s, is mostly unknown. Hardly any common but low-risk variants or rare, high-risk variants have been discovered. [Courtesy of Jose Bras, Rita Guerreiro, UCL.]
What does this current information imply for the genetic architecture of DLB? This question is often addressed with a diagram that depicts the risk a given variant confers over how frequently it occurs in a population. The most frequent variants confer small amounts of risk—unsurprisingly, because otherwise much of the population would have the disease. The highest risk comes from rare, autosomal-dominant mutations. For both AD and PD, dozens of low-risk variants are known, as are smaller numbers of highly penetrant variants. Some genes, such as SNCA, can show up in both corners, once as a triplication causing rare, early onset disease and once as a common variant contributing minimally to a person’s overall genetic risk. The middle space features APOE and TREM2 for AD and GBA and LRRK2 for PD, but largely awaits findings from ongoing deep-sequencing initiatives. In DLB, Bras said, this diagram remains an almost empty canvas, except for a few initial dots previously known from AD and PD. “So far we have not yet identified any genes specific for DLB. All genes we know to be involved in DLB are also involved in AD or PD. The work we are doing now will allow us to improve this,” Bras said.
“This data certainly suggests that there is something about the genetics of DLB that will be different than AD or PD,” said Bradley Boeve of the Mayo Clinic in Rochester, Minnesota. Scientists are waiting to see if that “something” is a particular multigenic predisposition, or a gene unique to DLB, or perhaps variants that drive the disease-specific topography of a given pathogenic protein.
In Fort Lauderdale, Cyrus Zabetian of the University of Washington, Seattle, presented a comparison of candidate genes in a smaller sample of 348 neuropathologically confirmed DLB cases and 102 PDD cases from seven centers in the United States. Finding broadly similar results, this study suggested that ApoE4 is more common in DLB, the disease that is arguably closer to AD, than in PDD, the disease nearer to PD. “It was striking that homozygous carriers of ApoE4 were greatly overrepresented among DLB cases,” Zabetian said. Au contraire, pathogenic variants of GBA were more common in the PDD patients than the DLB patients, offering a genetic hint of where those genes lie along the Lewy body diseases spectrum that spans from AD to DLB to PDD to PD. “If you carry ApoE4, it pushes you toward an earlier presentation of dementia, hence you are more likely to be classified as having DLB. GBA mutations, on the other hand, push you toward α-synucleinopathy, so you are more likely to be classified as having PDD,” Zabetian said.
Another hint that ApoE4 puts its finger firmly on the AD end of the DLB scale came from a study relating ApoE genotype to neuropathology in large series of people who had had a diagnosis of DLB during life. Melissa Murray from the Mayo Clinic in Jacksonville, Florida, reported that while ApoE4 carriers tended to have worse α-synuclein pathology than non-carriers, that link was due to the presence of amyloid pathology. About 90 percent of patients with DLB have AD pathology, as well, (see Part 3 of this series). In AD research, ApoE4 is increasingly seen as an indicator that amyloid is present in the brain with age, and several presentations in Fort Lauderdale, including Murray’s, pointed in the same direction for DLB.
At the conference, some scientists voiced doubt that genetics would ever be as instructive in DLB as it has become in frontotemporal dementia, in part because few large pedigrees are known. “In the clinic, by and large DLB is not an inherited disorder,” agreed Ian McKeith of Newcastle University, U.K. At the same time, several affected people who had come for the concurrent patient and care-partner track of this conference brought up a family pattern of DLB. They asked the scientists to explain their possible genetic versus environmental risk, for example among farming families in whom adult sons and cousins are wondering if handling the same pesticides as had their affected father and uncles will predispose them to a Lewy body disorder. Asked like this, the scientists had to answer simply, “We do not know.”—Gabrielle Strobel
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K.
alpha-Synuclein locus triplication causes Parkinson's disease.
Science. 2003 Oct 31;302(5646):841.
PubMed.
Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, Buchman AS, Larson EB, Crane PK, Kaye JA, Kramer P, Woltjer R, Kukull W, Nelson PT, Jicha GA, Neltner JH, Galasko D, Masliah E, Trojanowski JQ, Schellenberg GD, Yearout D, Huston H, Fritts-Penniman A, Mata IF, Wan JY, Edwards KL, Montine TJ, Zabetian CP.
GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology.
Neurology. 2012 Nov 6;79(19):1944-50.
PubMed.
Nalls MA, Bras J, Hernandez DG, Keller MF, Majounie E, Renton AE, Saad M, Jansen I, Guerreiro R, Lubbe S, Plagnol V, Gibbs JR, Schulte C, Pankratz N, Sutherland M, Bertram L, Lill CM, DeStefano AL, Faroud T, Eriksson N, Tung JY, Edsall C, Nichols N, Brooks J, Arepalli S, Pliner H, Letson C, Heutink P, Martinez M, Gasser T, Traynor BJ, Wood N, Hardy J, Singleton AB, International Parkinson's Disease Genomics Consortium (IPDGC), Parkinson's Disease meta-analysis consortium.
NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases.
Neurobiol Aging. 2015 Mar;36(3):1605.e7-12. Epub 2014 Aug 4
PubMed.
Guerreiro R, Escott-Price V, Darwent L, Parkkinen L, Ansorge O, Hernandez DG, Nalls MA, Clark L, Honig L, Marder K, van der Flier W, Holstege H, Louwersheimer E, Lemstra A, Scheltens P, Rogaeva E, St George-Hyslop P, Londos E, Zetterberg H, Ortega-Cubero S, Pastor P, Ferman TJ, Graff-Radford NR, Ross OA, Barber I, Braae A, Brown K, Morgan K, Maetzler W, Berg D, Troakes C, Al-Sarraj S, Lashley T, Compta Y, Revesz T, Lees A, Cairns NJ, Halliday GM, Mann D, Pickering-Brown S, Powell J, Lunnon K, Lupton MK, International Parkinson's Disease Genomics Consortium, Dickson D, Hardy J, Singleton A, Bras J.
Genome-wide analysis of genetic correlation in dementia with Lewy bodies, Parkinson's and Alzheimer's diseases.
Neurobiol Aging. 2016 Feb;38:214.e7-10. Epub 2015 Nov 2
PubMed.
Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, Escott-Price V, Hernandez DG, Nalls MA, Clark LN, Honig LS, Marder K, Van Der Flier WM, Lemstra A, Scheltens P, Rogaeva E, St George-Hyslop P, Londos E, Zetterberg H, Ortega-Cubero S, Pastor P, Ferman TJ, Graff-Radford NR, Ross OA, Barber I, Braae A, Brown K, Morgan K, Maetzler W, Berg D, Troakes C, Al-Sarraj S, Lashley T, Compta Y, Revesz T, Lees A, Cairns N, Halliday GM, Mann D, Pickering-Brown S, Dickson DW, Singleton A, Hardy J.
Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies.
Hum Mol Genet. 2014 Dec 1;23(23):6139-46. Epub 2014 Jun 27
PubMed.
Gan-Or Z, Mirelman A, Postuma RB, Arnulf I, Bar-Shira A, Dauvilliers Y, Desautels A, Gagnon JF, Leblond CS, Frauscher B, Alcalay RN, Saunders-Pullman R, Bressman SB, Marder K, Monaca C, Högl B, Orr-Urtreger A, Dion PA, Montplaisir JY, Giladi N, Rouleau GA.
GBA mutations are associated with Rapid Eye Movement Sleep Behavior Disorder.
Ann Clin Transl Neurol. 2015 Sep;2(9):941-5. Epub 2015 Jul 31
PubMed.


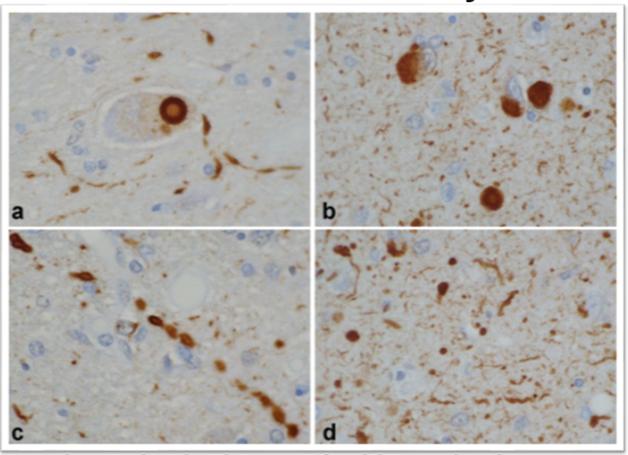
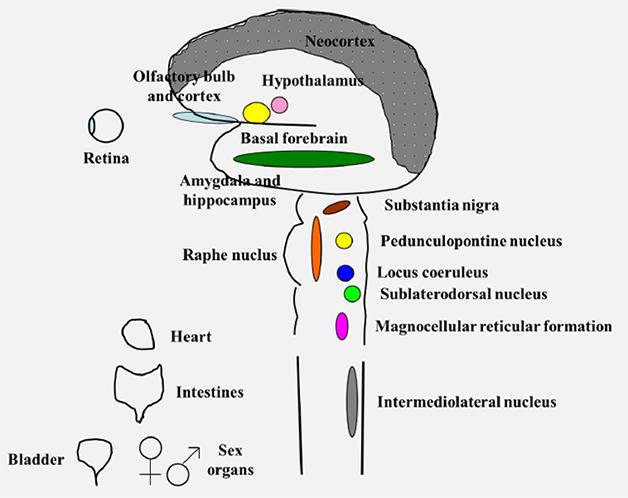



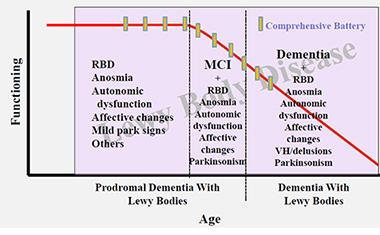



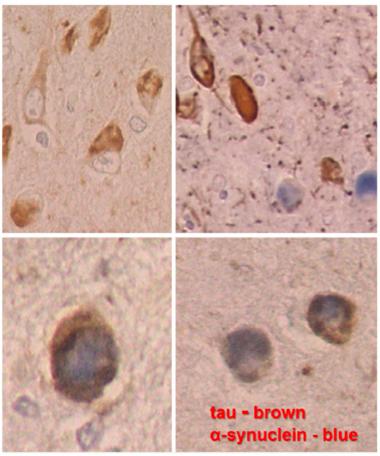
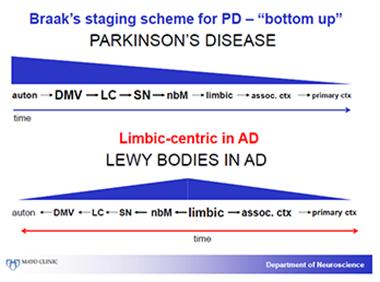


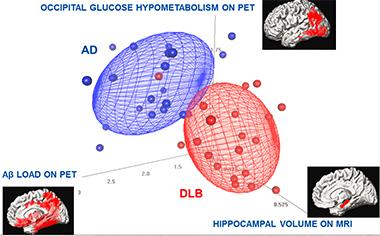

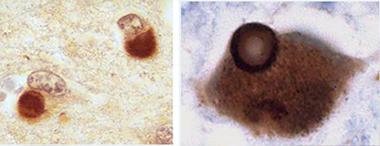
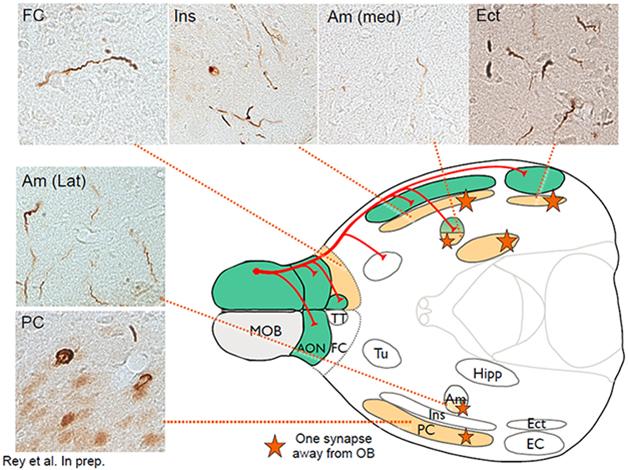
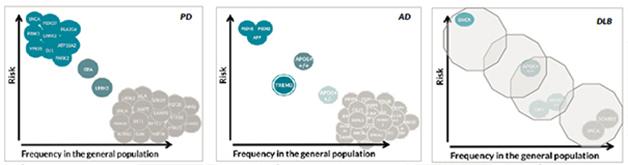
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.