Dementia with Lewy Bodies: Sharper Image for a Formerly Blurry Disease
Quick Links
Dementia with Lewy bodies is a common α-synucleinopathy that blends dementia with parkinsonism and psychiatric illness. When scientists met on December 1-4 in Fort Lauderdale, Florida, for the International Dementia with Lewy Bodies Conference, the strength of Japanese data on diagnostic MIBG scintigraphy was the big surprise (see Part 2 of this series). Even so, interspersed throughout the meeting were many more incremental advances. Researchers are deploying new methods to tease apart the overlap with Alzheimer’s and Parkinson’s that has long bedeviled DLB research. Together, the insights are converging to give definition to a disease that has long struggled for recognition. This more distinct identity of DLB emerges as scientists better understand which underlying protein pathology gives rise to which of aspect of its symptoms, or to how fast DLB progresses.
Importantly, researchers are increasingly relying on imaging data and tissue from people who have been studied in life and then received a definitive neuropathological diagnosis after death. Indeed, both imaging and cerebrospinal fluid (CSF) data presented at the Fort Lauderdale conference were anchored by neuropathology. This reflects an effort to understand the heterogeneity of DLB, a disease driven by accumulation of at least three toxic proteins—α-synuclein, tau and Aβ—in different regions of the brain, at different times, in different people. A major push in the DLB field goes toward accounting for what each pathological protein contributes to the disease during life.
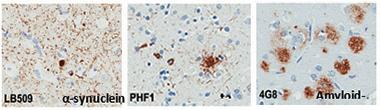
Three Villains.
Protein deposits of α-synuclein, tau, and Aβ plague the brains of people with DLB. [Courtesy of Tanis Ferman, Mayo Clinic.]
Dennis Dickson of the Mayo Clinic in Jacksonville, Florida, set the neuropathologic stage. He first aligned Braak’s staging of α-synuclein pathology in Parkinson’s disease—spreading from the bottom up via the vagus nerve and pons through the midbrain, basal forebrain, and eventually throughout the cortex—with categories proposed by Kenji Kosaka of Yokohama City University, Japan. (Kosaka started describing cases in 1976, and several researchers in Fort Lauderdale said that alongside the eponymous Parkinson’s and Alzheimer’s diseases, DLB might fairly have been named Kosaka’s disease).
In 1980 Kosaka had proposed three categories. The first, a restricted brainstem type, corresponds to Braak stages 1 to 3; the second, a transitional type with pathology in the basal forebrain, corresponds to Braak stage 4; the third, a diffuse type with Lewy bodies in the cortex, corresponds to Braak stages 5 and 6. Do the patients who walk into the clinician’s door fit these pathological bins, Dickson asked? Not so neatly.
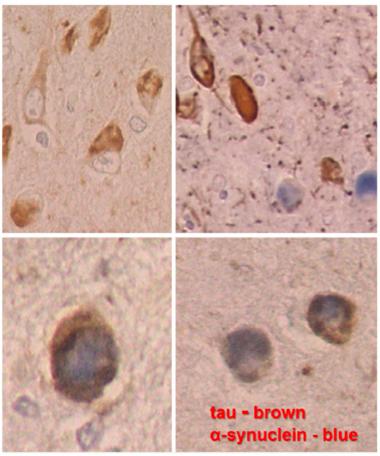
In DLB, neurons in the amygdala contain both aggregated tau (brown) and α-synuclein Lewy bodies (blue). Sometimes both co-localize in the same neuron (higher magnification at bottom). [Courtesy of Dennis Dickson, Mayo Clinic.]
First off, the most common Lewy body disease is Alzheimer’s. Fully a fifth of AD cases have ample α-synuclein pathology, Dickson said. They have it in the amygdala—just as in Braak stage 4 or Kosaka transitional Lewy body disease. Their α-synuclein pathology there occurs side by side with Alzheimer’s tau pathology. “The amygdala is uniquely vulnerable to both processes,” Dickson said, while showing images of α-synuclein and tau filaments mingling even in the same neurons in the amygdala. Secondly, the olfactory bulb is also highly prone to this α-synuclein/tau double whammy, often in the same people who have it in the amygdala. The brainstem, which is emblematic of Parkinson’s, tends to be unaffected in these olfactory/amygdala cases of DLB. The amygdala and olfactory bulb are part of the brain’s limbic system.
In essence, Dickson said, Braak’s bottom-up PD staging system works largely as advertised for PD, but not for DLB. In that disease, many cases have most α-synuclein pathology in the limbic system, and it spreads from there up into both the association and primary visual cortex, and down to the autonomic system.
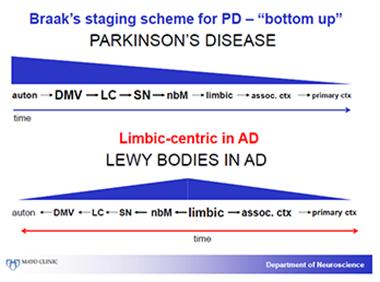
The distribution of α-synuclein pathology in Parkinson’s differs from that of DLB. In DLB, the limbic system predominates and appears to be affected early. [Courtesy of Dennis Dickson, Mayo Clinic.]
How do DLB cases shake out when amyloid pathology is added in? Seeing the patterns of three age-related proteopathies gets even more complicated. Dickson attempted to do this by plotting both tau pathology as per Braak stages and amyloid pathology as per Dietmar Thal phases against α-synuclein pathology as per Kosaka.
He did this with a Mayo Clinic series of 808 people who had had either AD, DLB, PDD, or PD. This created a complicated matrix of the bewildering variability of these diseases. Still, some themes emerged:
- In people who present clinically as DLB, pure Lewy body disease without AD pathology is rare. Nine out of 10 also have tangles and plaques, and six in 10 have a high burden of plaques.
- Among people diagnosed with PDD, i.e., who develop parkinsonism and later also dementia, one in five have a pure α-synuclein disease. The rest have both synuclein and AD pathology, and many have a heavy amyloid burden. This disease is extremely variable, Dickson said.
- Among people who present with Parkinson’s, four in 10 have no AD pathology; the remaining six have mostly a low burden of it.
This means that Lewy pathology in the cerebral cortex, particularly its limbic region, is common in DLB but minimal in Parkinson’s. In DLB, the olfactory bulb and amygdala are more prone to α-synuclein pathology than in PD. Because Lewy body disorders afflict old people, they frequently occur together with Alzheimer’s pathology, Dickson said. When their AD pathology is severe, the clinical balance of their symptoms tips toward AD. “The neuropathological diagnosis of DLB is a probability statement that takes into account the severity of both Lewy and Alzheimer pathology,” Dickson said.
You, Too, Tau?
Several speakers reported converging data on the question of what the accompanying tau tangles might be doing to a person who is living with DLB. In short, tau seems to speed up their decline. Tanis Ferman, a neuropsychologist at Mayo in Jacksonville who follows patients over time, wondered why some people with DLB decline gradually while others drop off precipitously. Analyzing neuropathology against clinical data of people who had died with DLB, Ferman first noticed that the clinical picture of DLB arose both in patients with transitional LBD, i.e., Kosaka’s more restricted distribution of α-synuclein, and with diffuse LBD, where Lewy bodies are spread throughout the cortex. In other words, cortical Lewy bodies were not necessary to make people symptomatic. That said, people with both diffuse LBD and with the most tau changed the most over time. “People with the greatest tau and synuclein burden overall declined fastest,” Ferman said.

How much pathology a person has determines how fast their disease gets worse. [Courtesy of Tanis Ferman, Mayo Clinic.]
In a second, larger sample, Ferman split transitional and diffuse cases into high and low tau burden. This unmasked more differences. People with a lot of tauopathy in addition to their synucleinopathy not only progressed fastest, they also had worse memory loss and were likelier to carry ApoE4 and be female than the DLB patients with a low tau burden. Also, they were less likely to have REM sleep behavior disorder (RBD). In essence, Ferman saw facets of DLB’s AD component via the tau pathology. (Amyloid plaques were tightly correlated with tangles in this study, hence the “high tau” cases also had high Aβ.) The polar ends of the spectrum support this, as AD is more common in women and ApoE4 carriers, and marked by memory decline, whereas PD is more common in men, with more REM sleep disorder but intact memory, and lower frequency of ApoE4. The fastest decline, said Ferman, results from having a high burden of diffuse α-synuclein deposits paired with a high burden of AD pathology.

In this sample, people with both diffuse Lewy body disease—the most severe Kosaka category of synucleinopathy—and lots of tangles declined most rapidly on a measure of cognition (red). [Courtesy of Tanis Ferman, Mayo Clinic.]
Can multimodal imaging help in the face of this complex underlying burden of mixed proteopathy? In the absence of MIBG cardiac scintigraphy, a single imaging marker is not enough to distinguish the AD and DLB syndromes clearly from each other. Researchers hope that seeing several aspects of the disease at once will make things clearer. Kejal Kantarci of the Mayo clinic in Rochester, Minnesota, is analyzing a host of imaging findings from a cohort of DLB patients who have since been autopsied. In Fort Lauderdale, Kantarci reported that, at least in her hands, multiple markers can almost completely separate the two syndromes.
Kantarci also reported that those DLB patients who turned out to have had abundant concomitant AD pathology are the ones whose medial temporal lobe (MTL) shrank in life. Very little MTL atrophy registers in group studies of DLB, but it does show up in individuals who have lots of plaques and tangles. “The higher the Braak stage, the more hippocampal loss,” Kantarci said. Hippocampal atrophy in AD is widely thought to reflect tangle pathology.
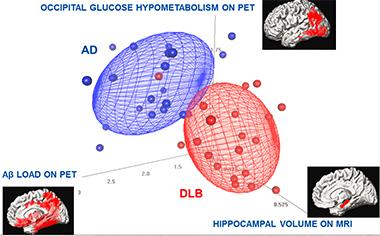
In this study of 21 patients, each with DLB or AD, three imaging methods combined distinguished the two better than each individual scan. Except for one DLB patient, all AD and DLB patients are separated using multimodality imaging measures.
With that shrinkage came shorter survival. Jonathan Graff-Radford, also at Mayo Rochester, reported that in a cohort of 167 DLB patients, those with the smallest hippocampal volume at baseline died soonest after the disease was diagnosed. Graff-Radford’s model calculated a difference in predicted survival of almost four years between the DLB patients who had a shrinking hippocampus and carried ApoE4, a genetic indicator of Aβ pathology, and those DLB patients who had a relatively larger hippocampus and no ApoE4 allele. The former could be seen to have more AD admixed into their DLB, the latter less.
Yet another hint that AD pathology worsens DLB came from multimodal imaging by Stephen Gomperts of Massachusetts General Hospital, Boston. Gomperts confirmed that AD pathology has real repercussions for progression when it co-exists with Lewy pathology. He follows patients with the overlapping synucleinopathies DLB, PDD, and PD who are getting both PiB amyloid and T807/AV1451 tau PET scans. Thus far, he sees amyloid positivity in most people with DLB. Much like Ferman saw based on neuropathology, Gomperts sees that when a person’s PiB uptake, i.e., their amyloid burden, is high, then that person’s cognition will decline faster on the CDR-sb and the MMSE. Likewise, in the people with Parkinson’s who have a positive amyloid scan, high uptake predicts faster decline from normal cognition to MCI, and from MCI to PD dementia. In contrast, amyloid did not change how fast parkinsonian motor symptoms declined, Gomperts said.
Gomperts also described an early experience with tau PET, in a cohort of 29 controls, seven people with DLB, eight with PD-MCI, and nine with cognitively normal PD. Gomperts sees the most T807 retention in people with DLB. Their signal in the inferior temporal gyrus can approach levels published for AD (Johnson et al., 2015). Gomperts noted that this signal is highly variable in the DLB patients he has scanned thus far. In contrast, he saw a weaker tau signal in the inferior temporal gyrus of PDD, PD, and controls.
Those DLB patients who are the most cognitively impaired on CDR-sb and MMSE have the most T807 retention. When Gomperts added a cortical thickness measure from MRI scans into this model, he saw a three-way interaction. In essence, as T807 retention went up and the cortex thinned out, people were more cognitively impaired.
This data was well received, though Kantarci noted that in her hands, the T807 signal imperfectly matches postmortem pathology in terms of both location and amount. She believes T807 may ignore certain kinds of tau pathology in DLB. With regard to non-specific binding of T807, Gomperts pointed out that a recently published validation study (Marquie et al., 2015) showed that T807 can also bind neuromelanin in the substantia nigra as well as certain blood products; however, this is based on putting T807 directly onto brain tissue slices, not yet on a neuropathological confirmation of people who had had scans in life.
Beyond clinico-pathological and imaging work, fluid biomarkers now also confirm that AD pathology speeds up DLB. CSF biomarkers can track DLB from the beginning, while neuropathology only gets one look at the brain in the aftermath of disease. Unlike PET, CSF tests can measure multiple proteins at the same time, and they cost less than imaging, making them well-suited for repeat assessment in large cohort studies. In Fort Lauderdale, Evelien Lemstra of VU Amsterdam reported the first results from an ongoing study of one of the largest cohorts of DLB patients in Europe. Before she showed her data, Lemstra tried to soften the opposition to lumbar puncture that is ingrained at many sites in North America by saying, “We just finished CSF collection in 1,000 patients and see a very low rate of headache or other side effects.”
CSF Aβ and tau studies in AD are well-established (see the Alzbiomarkers database for meta-analyses). As for DLB, Lemstra reported that among 111 patients with clinically probable DLB, 69 had the CSF AD profile of a low tau/Aβ42 ratio (see Duits et al., 2014). They were more likely to carry an ApoE4 allele and less likely to have a positive DaTscan than the 42 DLB patients who did not have the CSF AD signature, Lemstra said.
The DLB patients with AD CSF markers had more severe hallucinations and delusions. They performed worse on tests of memory, but better on tests of executive or visuospatial function, attention, or language. Lemstra has insufficient follow-up data thus far to see if cognition in the two groups declines at different speeds, but she does see a trend that DLB patients with AD CSF markers get placed in nursing homes faster and die sooner. “The AD pathology in DLB is clinically relevant, and can be detected antemortem with CSF,” Lemstra told the audience. This cohort is part of the Europe-wide E-DLB project (see Part 6 of this series).
Given this convergence of evidence about the presence of AD pathology in DLB, and its contribution to the real-life experience of this disease, researchers in Fort Lauderdale called for more data sharing to confirm the importance of these insights in larger samples across the field. Others said the growing evidence already argues for evaluating anti-amyloid and anti-tau drugs in people with DLB who are confirmed at enrollment to have concomitant AD pathology.—Gabrielle Strobel
References
News Citations
Paper Citations
- Marquié M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, Klunk WE, Mathis CA, Ikonomovic MD, Debnath ML, Vasdev N, Dickerson BC, Gomperts SN, Growdon JH, Johnson KA, Frosch MP, Hyman BT, Gómez-Isla T. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015 Nov;78(5):787-800. Epub 2015 Sep 25 PubMed.
- Duits FH, Teunissen CE, Bouwman FH, Visser PJ, Mattsson N, Zetterberg H, Blennow K, Hansson O, Minthon L, Andreasen N, Marcusson J, Wallin A, Rikkert MO, Tsolaki M, Parnetti L, Herukka SK, Hampel H, De Leon MJ, Schröder J, Aarsland D, Blankenstein MA, Scheltens P, van der Flier WM. The cerebrospinal fluid "Alzheimer profile": easily said, but what does it mean?. Alzheimers Dement. 2014 Nov;10(6):713-723.e2. Epub 2014 Apr 8 PubMed.
Other Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.