More than 31,000 people attended the annual Society for Neuroscience meeting in Washington, D.C., November 15 to 19. Twenty-five symposia, 26 minisymposia, 101 nanosymposia, 667 poster sessions, and numerous lectures, workshops, and satellite events attracted researchers engaged in all manners of neuroscience. If you failed to soak it all in, or if you opted for the relative calm of the lab this year, then check out Alzforum's conference series.
Society for Neuroscience Annual Meeting: A Haven for Basic Science
The 31,000-plus scientists who descended on the U.S. capital for the Society for Neuroscience annual meeting, November 15-19, made for an enthusiastic crowd, even though there were fewer Alzheimer’s sessions than in previous years. From nifty new tools (read: Aβ electrode) to potential paradigm shifts (read: provenance of plaque microglia), Alzforum reporters will bring you highlights over the coming weeks. One announcement at the meeting was that Michela Gallagher of Johns Hopkins University, Baltimore, won the 2014 Mika Salpeter Lifetime Achievement Award. The society bestows this honor on leaders who have also promoted the advancement of female neuroscientists. Gallagher studies the effect of aging on cognition and, among other findings, discovered that overactivation of the hippocampus may cause people to forget, rather than remember (see May 2012 research news).
A Glial Tracer.
GE180 lights up the brain (circled) in a mouse model of AD. While the tracer is also taken up outside of the brain, researchers believe it could be used to track glial activation during disease. [Image courtesy of Cynthia Lemere, Brigham and Women's Hospital, Boston.]
Perhaps the biggest surprise at this year's meeting, a "game changer" to some scientists, came from studies of TREM2. This glial receptor made a splash two years ago when geneticists reported that a variant increased the risk for AD by around threefold. At SfN, researchers claimed that TREM2-positive cells that hover around plaques in the brain are not brain-resident microglia at all, but are in fact peripheral monocytes. If confirmed, this would represent a departure from what many in the field have assumed—namely that the glia mopping up Aβ plaques are brain-derived.
In related glial news, GE180, a PET ligand for tracking microglial activation, made its mark. Like other glial tracers that have been developed, GE180 binds a mitochondrial transporter protein called TSPO. Unlike the prototype TSPO ligand PK11195, however, GE180's PET signal comes from fluorine-18, which is more suitable for imaging, given its longer half-life. As a microglial tracer, GE180 appears to be more specific and selective than its predecessors, and researchers at the meeting were excited by the possibility of finally having a glial tracer that might work well in people. The tracer has been tested preclinically. Researchers reported that in a mouse model of AD, an experimental therapeutic returned GE180 uptake in the brain to baseline levels within three months.
In another technological advance, scientists at Washington University, St. Louis, have developed an Aβ electrode (see image below) that can measure changes in the concentration of the peptide in the brain over a 30-second time span, allowing researchers to more accurately track Aβ dynamics. The technology uses an Aβ antibody-tipped electrode. It could be adapted to measure other proteins, including tau, and potentially different forms of Aβ, though for now it appears to detect only monomers. Researchers at the meeting were intrigued at the possible uses for such electrodes. In other Aβ developments, researchers from CogRx, a small company in Pittsburgh, reported new compounds that attenuate the peptide's toxicity. They displace various forms of Aβ, including oligomers isolated from patient brains, from the cell surface, and CogRx has identified sigma-2/progesterone receptor membrane component 1 as a specific binding receptor for the compounds. The company plans to enter compounds into human trials early next year. In the "out of left field" category, scientists from Revalesio, a small company in Tacoma, Washington, reported that "nanobubbles," positively charged spheres laden with oxygen, improve pathology and behavior in the 5xFAD mouse model of AD. Before eyes roll, dear reader, get the full story to judge for yourself whether the science behind these anti-inflammatory bubbles seems solid. They are in clinical testing for a variety of conditions.
Micro-immunoelectrode.
This sliver of an electrode measures Aβ in the interstitial fluid twice per minute, offering a high-resolution view of the peptide’s changes. [Image courtesy of John Cirrito, Washington University.]
More unusual receptors made the news at SfN. Researchers reported that the P2Y1 subtype of ATP purinergic receptor in astrocytes promotes hyperactivation of these cells around plaques, a phenomenon that has been observed for decades. While P2YI turns on early in AD, perhaps even before plaques appear, an inhibitor of the receptor restored normal astrocytic activity in transgenic APP mice. Another purinergic receptor, the A2A adenosine receptor, has also been implicated in the disease. At SfN, researchers reported that in people with AD, and in transgenic mouse models, astrocytes overexpressed this receptor. A2A levels correlated with plaque burden and disease progression. Genetically removing these receptors from astrocytes in APP transgenic mice improved their memory, while activating them did the opposite. The findings suggest that the A2A purine receptor in astrocytes contributes to memory loss in AD.
Calcium dysregulation may be another early casualty of the disease. Researchers know that expression of endoplasmic reticulum ryanodine channels, and calcium signaling through these channels, goes up in AD-transgenic mice before they develop memory deficits. New data at SfN suggested that preventing this calcium dyshomeostasis preserves hippocampal synapses and spines, and restores synaptic plasticity. This made researchers think that targeting the ryanodine receptor might be worth trying in AD patients. Compounds that restore calcium homeostasis and RyR-signaling in hippocampal slice preparations and in AD mice in vivo are prototypes for potential drug discovery.
There was plenty of basic biology on display at SfN as well. Exosomes were a hot topic in a special lecture and mini- and nanosymposia, including one co-chaired by Xandra Breakefield from Massachusetts General Hospital, Boston, who won the 2013 Mika Salpeter Award. Scientists reported new ways in which these small extracellular vesicles function in cell-to-cell communication in the nervous system. New evidence suggests that cells specially prepare and package protein and RNA into exosomes to be delivered to other cells, where they affect axon growth and cell survival. Brain tumor cells may use them to prime the environment for their growth. Exosomes may also conspire in the spread of proteins involved in neurodegenerative disease. Evidence from stem cells suggests that neuronal exosomes spread Aβ and α-synuclein, while another study implicated microglial exosomes in the spread of tau. In addition, studies hinted that amyloid precursor protein and the secretases necessary for Aβ production can be enveloped in exosomes, hinting that the peptide might be produced in these vesicles outside the cell. Neurons from Down’s syndrome patients expel more exosomes than do control neurons, which may partly explain why these people accumulate so much Aβ in their brain. Scientists at the meeting wondered if these mechanisms could offer new targets for preventing the spread of pathogenic proteins around the central nervous system.
In genetics news, researchers at Genentech reported a rare coding mutation that associates with AD in two families affected by late-onset disease. They corroborated the finding in four case/control cohorts. The mutation lies in the UNC5C gene; it encodes a Netrin receptor that directs axon migration during development. Since the mutation doubles risk for AD, making it one of the strongest risk factors after ApoE4 and TREM2, researchers at the conference hailed the finding as potentially important. In vitro data suggests this mutation increases susceptibility to Aβ toxicity, causing cell death.
In other genetics reports, researchers claimed that a mutation in Tmp21, a modulator of γ-secretase cleavage, associates with AD, while calling a mutation that decreases expression of CD33 protective. In the search for such protective variants, many groups have looked to older people with ApoE4 who seem to defy their odds and live free of dementia past age 75. At SfN, researchers reported finding protective variants in genes previously associated with AD in a cohort of 24 people who are homozygous for ApoE4. They also spotted potentially protective mutations in genes involved in pathways that have been implicated in the disease.—Tom Fagan and Gwyneth Zakaib
For more than a decade, scientists have used microdialysis to estimate how the concentration of Aβ in the brain’s interstitial fluid changes in response to synaptic activity. Unfortunately, the technique allows sampling every half-hour at most, but neurons fire much more frequently. “We were orders of magnitude off the timescale for Aβ generation and release,” said John Cirrito, Washington University in St. Louis. Cirrito spoke to an audience at the Society for Neuroscience annual meeting, held in Washington, D.C., November 15 to 19. To close that gap, Cirrito’s group has developed a carbon fiber electrode that measures the concentration of Aβ every 30 seconds. This micro-immunoelectrode specifically binds to Aβ and oxidizes the peptide’s lone tyrosine, detecting the electrons released as current. Using this new technique, scientists will be able to observe up-to-the-minute Aβ fluctuations.
Protruding from a glass pipette, this carbon fiber electrode is 5 μm in diameter. It can rapidly detect fluctuations in Aβ. [Image courtesy of John Cirrito.]
In regular microdialysis, researchers insert a 2-mm long, 300-μm diameter probe into the brain, where it collects Aβ that crosses its semi-permeable membrane. An ELISA then measures the concentration of Aβ every 30 to 60 minutes (see Cirrito et al., 2003). When it debuted, this technique gave scientists their first opportunity to measure the concentration of Aβ over time in the same mouse. When working at David Holtzman's lab at WashU, Cirrito and colleagues found that, on average, Aβ production rose when synapses were excited, and fell when they were quieter (see Dec 2005 news story).
A few years later, Cirrito caught a presentation by Chenzhong Li, Florida International University, Miami, about an antibody-tipped microelectrode that could rapidly detect vascular endothelial growth factor coming from tumor cells (see Prabhulkar et al., 2009). After visiting Li’s lab, Cirrito realized that the same technique could speed up detection of Aβ.
Together, Li and Cirrito developed a 5-μm diameter micro-immunoelectrode that varies between 20 and 50 μm in length (see image above). It is five orders of magnitude smaller than the probe used in microdialysis, about half the size of a neuron. A previous publication described its first use in vitro (see Prabhulkar et al., 2012). Carla Yuede, who works in Cirrito's lab, helped adapt it for use in vivo. Inserted into the hippocampus of an anesthetized APP/PS1 transgenic mouse, the microelectrode, charged to 0.65 volts, oxidized the tyrosine residue on the human Aβ, capturing the electrons released. “The amount of current generated is directly proportional to the number of tyrosines that bind the electrode,” said Cirrito. Because rodent Aβ lacks this tyrosine, the micro-immunoelectrode picks up only human Aβ, he told Alzforum.
To ensure that the electrode detects Aβ specifically, the tip is coated with an anti-Aβ antibody, and any remaining spaces are filled in with bovine serum albumin (see image below). The researchers can substitute different antibodies, depending on whether they want to detect Aβ40 or Aβ42. While these antibodies should be highly specific, they also need to have a reasonably low affinity for Aβ, so as not to bind the peptide too tightly and saturate the electrode immediately. Even so, a given electrode only lasts about 90 minutes. Cirrito said he is still working to figure out why they eventually give out.
So far, Cirrito’s group has conducted initial experiments that refine previous estimates about Aβ production and synaptic activity. For instance, when the researchers injected anesthetized APP/PS1 mice with picrotoxin to block inhibitory GABAergic signaling, Aβ levels rose gradually between three and 15 minutes later, and then stabilized out to 90 minutes, the lifetime of the electrode. Before, the researchers only knew that levels rose, on average, within an hour. In another experiment, Cirrito and colleagues blocked Aβ production with a γ-secretase inhibitor. They calculated that half the Aβ in the interstitial fluid cleared in 40 minutes, slightly faster than the 54-minute half-life estimated from microdialysis.
Aβ Only, Please.
Antibodies that coat the tip of this micro-immunoelectrode ensure that electrons from oxidized tyrosine originate solely from Aβ, not other proteins (blue). [Image courtesy of John Cirrito.]
On her SfN poster, Yuede further broke down the clearance measurements. She found that when mice were injected with a γ-secretase inhibitor, Aβ dropped in two phases: for the first 15 minutes, the peptide cleared rapidly, but for the next hour, disappeared at a slower rate. The researchers predict that these two phases correspond to different mechanisms of clearance from the brain. For instance, when Yuede blocked p-glycoprotein, a protein thought to pump Aβ through the blood-brain barrier, the first phase stayed the same, but the second was slower, suggesting that p-glycoprotein handles the slower clearance mechanism. “We do not know yet what is responsible for the faster clearance,” Cirrito told Alzforum. Since research suggests that people with Alzheimer’s disease clear Aβ more slowly from the brain (see Jul 2010 news story), clearance mechanisms may be important to target for therapeutics. “If you want to enhance clearance to help AD, you may want to prioritize one pathway over another,” said Cirrito.
This technique won’t replace microdialysis, said Cirrito, but it will give researchers another option if they want to measure rapid Aβ fluctuations. For example, previous research by Holtzman’s lab suggests that Aβ production wanes during sleep and ramps up again during wakefulness (see Kang et al., 2009). This technique will allow researchers to see how quickly those changes take place, and whether a lag phase follows sleeping or waking, Cirrito said.
Researchers praised this as a “fantastic” new methodology. “This will revolutionize the analysis of interstitial fluid levels of Aβ,” Gary Landreth, Case Western Reserve University, Cleveland, wrote to Alzforum. The real-time measurements it allows represent a vast improvement over the more cumbersome microdialysis. “One of the most compelling aspects of this work is that it should be applicable to other analytes which have an appropriately placed tyrosine and for which there is a good antibody,” wrote Landreth. Cirrito's group has already developed a prototype electrode that uses an anti-α-synuclein antibody, and researchers have expressed interest in developing it for tau. The release of both peptides may depend on synaptic activity (see Fortin et al., 2005; Yamada et al., 2014).
At Cirrito’s presentation, Miranda Reed, West Virginia University, asked whether different types of anesthesia could influence Aβ measurements from the micro-immunoelectrode. Cirrito responded that he had no comparative data to present yet, but expected it might. Sleep promotes glymphatic clearance of solutes, including Aβ, from the brain (see May 2014 news story). In the future, Cirrito hopes to perform measurements in awake mice and avoid problems with anesthesia altogether. In answer to another audience question, Cirrito explained that the 30-second time window for the electrode was necessary to enable enough Aβ to bind and give a readable signal, but not so long that the all the available antibodies would become laden with unoxidized Aβ.—Gwyneth Dickey Zakaib
Research into what microglia do in Alzheimer's disease got a boost when geneticists discovered that mutations in the glial cell surface receptor TREM2 increase the risk for AD. But is the problem really with the microglia? At the Society for Neuroscience annual meeting, held November 15 to 19 in Washington, D.C., researchers surprised the audience by claiming that TREM2-expressing cells in the vicinity of Aβ plaques in the brain are monocytes that came in from the blood. Even more surprising, the new research links reduced TREM2 to attenuation of amyloid plaque load in the hippocampus. "This could totally change our assumptions about Aβ clearance," said Gary Landreth, Case Western Reserve University, Cleveland, who directed the work along with Bruce Lamb at the Cleveland Clinic. Scientists who saw the presentations suggested cautious interpretation, noting previous studies suggesting that microglia make more TREM2 than peripheral macrophages do and that a loss of TREM2 function puts people at risk for neurodegenerative diseases, including Alzheimer’s.
The basis for the peripheral monocyte claim comes from flow cytometry analysis of cells expressing TREM2. Crystal Miller, from Lamb's lab, examined TREM2 in 5xFAD and APP/PS1 mouse models of AD, both of which aggressively develop amyloid plaques. She reported that as the mice aged, TREM2 increased in the brain and co-localized with Congo Red-positive Aβ plaques as judged by immunohistochemistry with a TREM2 antibody. She crossed a mouse with a Lac Z gene knocked into the TREM2 locus with the AD models and, when she looked at amyloid plaques in their brains, saw the characteristic blue staining that this bacterial enzyme generates in response to the sugar compound X-gal. This helped confirm that glia around plaques express TREM2. Miller told Alzforum that questions had been raised about the specificity of TREM2 antibodies and she wanted to ensure she was looking at TREM2, not some other epitope. When she knocked out the TREM2 gene she saw no expression of the protein, confirming the antibody’s specificity. Using immunohistochemistry, Miller also saw TREM2-positive cells around Aβ deposits in human brain tissue.
Monocytes surround plaques:
In APP/PS1 mice, CD45-positive cells (dark brown) surround Congo Red-positive plaques (left), but not if TREM2 is knocked out (right). [Image courtesy of Crystal Miller.]
What are the TREM2-positive cells crowding around amyloid plaques? They lacked the purinergic receptor P2RY12, a marker of microglia. Instead, they flaunted Iba1, a marker of pro-inflammatory, non-phagocytic cells. This contravenes dogma, Landreth told Alzforum. "The literature suggests that TREM2 is anti-inflammatory and pro-phagocytotic, but here we found plaques associating with TREM2-positive cells that must be pro-inflammatory and phagocytically inactive based on the vast Iba1 literature," he said.
To characterize the TREM2 cells further, Miller isolated them by flow cytometry, then analyzed their other markers. To her surprise, she found TREM2 in cells that highly expressed CD45. "This was a huge shock," said Miller. Scientists believe that cells flush with CD45 are peripheral monocytes, not brain-resident microglia. The TREM2-positive cells expressed other markers of peripheral cells as well, including F480 and Ly6C.
To Miller, the data suggest that TREM2-expressing cells in the brain must be recruited from the periphery. In support of this, she found four- to fivefold more TREM2-positive cells in the blood of 4-month-old APPPS1 mice than in the blood of wild-type controls. While this suggests that peripheral monocytes in AD mouse models make TREM2 while in the blood, Miller said that the cells might also begin to express the protein after they have infiltrated the brain.
Researchers at the meeting seemed puzzled by the findings. Dave Morgan, University of South Florida, Tampa, cautioned that Joseph El Khoury at Massachusetts General Hospital, Charlestown, and others have shown that microglia preferentially express TREM2 (see Thrash et al., 2009, and Hickman et al., 2013). At the same time, El Khoury's lab has implicated peripheral monocytes in clearing plaques (see Mar 2007 news story). "The microglial role just gets more interesting," said Morgan.
Others wondered if the findings might be specific to mice. Both Elliott Mufson from Rush University, Chicago, and Greg Cole of University of California, Los Angeles, noted that TREM2 expression ticks up only slightly in the AD brain, much less than in mouse models. Christian Haass, Ludwig-Maximilians-Universität, Munich, wondered if there might be something about these mice that prompts their peripheral cells to invade the brain. "Even how the animals are housed could be an important factor," he told Alzforum.
Monica Carson from the University of California, Riverside, who was not at the meeting but had heard about the findings, said the data get people to think about TREM2 biology. She said it was plausible that TREM2 cells might infiltrate the brain from the periphery. "We are starting to realize that the biology of TREM2 is not as simple as turning it on or off, or it being expressed in one cell rather than the other," she told Alzforum. Carson believes that the key to TREM2 expression in AD models is the milieu around plaques. "That environment might demand induction of TREM2 on whatever immune cell is there," she suggested. Carson previously reported that TREM2-positive cells around plaques are most likely brain-resident microglia (see Melchior et al., 2010). That was in the APP23 model, which lays down plaques more slowly than the 5xFAD and APP/PS1 models that Miller and colleagues used.
Carson also urged caution in interpreting TREM2 antibody interactions. Several groups have reported that TREM2’s ectodomain sheds from the cell surface and can bind to other nearby cells (see Hamerman et al., 2006, and Wunderlich et al., 2013). Antibodies that recognize that ectodomain may not distinguish between cells that express TREM2 and those that bind it, she said.
The origin of TREM2 cells aside, what effect does loss of the protein have on plaques? Taylor Jay, a graduate student in the labs of both Landreth and Lamb, reported that far fewer glia surrounded plaques in TREM2 knockout APP/PS1 mice than in control APP/PS1 animals. In fact, the even spacing, or tiling, of microglia seen in a normal mouse brain was about the same in APP/PS1 TREM2 knockouts, suggesting that TREM2 is essential for recruiting cells to plaques. "There are very few plaque-associated microglia in the TREM2 knockouts," said Landreth. This fits with work by Jason Ulrich and colleagues at Washington University, St. Louis, who found fewer glia surrounding plaques in APP/PS1 animals that have only one copy of the TREM2 gene (see Jun 2014 news story). Jay also reported fewer astrocytes around plaques in APP/PS1 TREM2 knockouts compared with APP/PS1 controls. Inflammation was down as well, judging by reductions in pro-inflammatory interleukin 1b and IL6.
Corroborating Ulrich's finding, Jay found as many amyloid plaques in the cerebral cortex of the APP/PS1 TREM2 knockouts as in controls. Ulrich has since extended his observations to homozygous knockouts, and reported at SfN that they resemble the heterozygotes. In the latter, glial numbers were down by half, while in the homozygote knockouts they fell further, to 25 percent of controls. He said cortical plaque load was unchanged in either case.
Unexpectedly, while Jay found no change in plaque load in the cortex, she reported fewer plaques in the hippocampus in her APP/PS1/TREM2-/- mice. Immunohistochemistry using the 6E10 anti-Aβ antibody suggested more than a threefold reduction in plaques in 4-month-old animals. ELISA of brain lysates showed almost a twofold reduction in insoluble Aβ, as well. The tau antibodies AT8 and AT180 detected twofold less phospho-tau in the vicinity of plaques in the TREM2 knockouts. "We found these results very surprising based on predicted TREM2 function," said Jay. "If anything, we would have expected to see an increase in pathology in TREM2 knockouts."
Carson was not surprised by the drop in plaques. "That would be consistent with models of stroke, where we see decreased pathology in TREM2 knockouts," she said (see Sieber et al., 2013). She noted that TREM2 expression in the mouse brain is very high during early development, but falls considerably by the time the animals reach 3 months of age. "That suggests there is some risk to having high expression," she said.
The overall feeling at this meeting was that much more work needs to be done to understand the role of TREM2 in disease. Some scientists wondered if TREM2-positive cells were really coming from the periphery, or if brain-resident microglia were changing their phenotype to express markers typically considered peripheral. Others noted that any model for TREM2 function needs to explain not just pathology of AD, but of FTD, PD, and other neurodegenerative diseases genetically linked to TREM2, and suggested that clearance of dead and dying cells by microglia might be key. Haass, who presented his recent findings linking TREM2 mutations to compromised microglial phagocytosis, agreed (see Kleinberger et al., 2014, and Alzforum webinar). "We see that removal of debris and cells is abolished in TREM2 knockouts and this may be the best way to look at its function," he said. —Tom Fagan
Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C.
TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis.
Sci Transl Med. 2014 Jul 2;6(243):243ra86.
PubMed.
Meet GE180: A PET Ligand for Tracking Neuroinflammation?
Amyloid β—check. Tau—check. Microglia—? Researchers have long sought a tracer suitable for imaging the activation of these immune cells in the living brain. After some ho-hum attempts, scientists once again hope that a new compound finally might fit the bill. At the Society for Neuroscience annual meeting, held November 15-19 in Washington, D.C., researchers reported for the first time that they picked up the tracer in the brains of live Alzheimer's mouse models using micro PET scans. Transgenic AD mice took up more GE180 in their brain than did control animals. Mice treated with an experimental AD drug took up less of the tracer over time, suggesting that GE180 could possibly gauge responses to therapies that reduce the activation of microglia. Researchers at the meetings seemed impressed by the findings, but wanted to know more about the ligand’s receptor.
GE180, like many glial tracers before it, binds a mitochondrial translocator protein, TSPO for short. Many tissues of the body make large amounts of TSPO, but a healthy brain produces very little. Activated microglia, on the other hand, crank out TSPO, making it a potential marker of brain inflammation. Fortuitously, the protein has an affinity for certain chemicals. Several such carbon-11 compounds, including the prototypical ligand PK11195, have been investigated as microglial PET tracers, but as Cynthia Lemere of Brigham and Women's Hospital, Boston, explained in Washington, GE180 makes a more versatile tracer. It sports fluorine-18, which has a longer half-life than C-11 and need not be synthesized on site.
Earlier this year, researchers reported that GE180 has a better signal-to-noise ratio than PK11195 when picking up glial activation in a mouse model of ischemic stroke (see Boutin et al., 2014). Researchers in Finland tested the ligand in a rat model of acute inflammation, and their paper gave the GE compound an edge over PK1195 in assays of the compounds’ binding potential (see Dickens et al., 2014).
Glial Activation Live:
Wild-type mice take up GE180 in peripheral tissues but 26-month-old APP/PS1 mice also trap the tracer in their brains (oval). [Image courtesy of Cynthia Lemere, Brigham and Women's Hospital, Boston.]
How does GE180 fare in AD models? Bin Liu, a senior postdoc in Lemere's lab, tracked its uptake in the brains of APP/PS1 transgenic mice and controls. Four-month-old animals took up little. By 26 months, wild-type mice showed a noticeable uptick in tracer binding in the brain, suggesting an age-related increase in glial activation. Old APP/PS1 animals took up even more of the tracer, in keeping with pathology driving glial inflammation.
To pinpoint where GE180 binds, Liu co-registered PET images to MRI scans of the mouse brain. Both the cortex and hippocampus in the old APP/PS1 mice took up more tracer than did controls. Importantly, Liu correlated uptake seen by PET with tissue autoradiography done on 1 millimeter slabs of the brain. In thinner, fixed cryo-sections, GE180 binding correlated with increased plaque deposition, said Lemere, and the TSPO-positive cells tested positive for the microglial marker CD68.
During question time, scientists at the meeting wanted to know more about TSPO. Dave Morgan, University of South Florida in Tampa, implored colleagues to develop good antibodies to the protein for immunohistochemical work. "We've been labelling microglia for years but have never had a good marker for TSPO," Morgan said. Lemere replied that AbCam now has a good monoclonal antibody to the mitochondrial protein.
Others wondered how TSPO is regulated, and if its expression returns to baseline levels once inflammation resolves. In her talk, Michelle James, Stanford University, Palo Alto, California, showed just that in another mouse model of AD.
Tamping Down Inflammation:
Autoradiographs show uptake of GE180 in the cortex (Ctx), hippocampus (HC), and choroid plexus (CP) of APP-transgenic mice is much reduced after three months on an experimental drug (right). [Image courtesy of Michelle James, Stanford University.]
James used micro PET to track GE180 uptake in mice expressing human APP with the London and Swedish mutations. To see if the compound can trace both the rise and fall of glial inflammation, James treated the animals with an experimental drug being developed by Frank Longo at Stanford and Stephen Massa at the University of California, San Francisco. LM11A-31 binds to the p75 neurotrophin receptor and PharmatrophiX, a company founded by Longo, recently completed a Phase 1 trial (see Nov 2013 conference story). Longo told Alzforum that a Phase 2a trial in AD patients is being planned.
James measured GE180 uptake in 5.5- to 7-month-old APP and wild-type mice to get a baseline scan. Nadia Belichenko at Longo's lab then administered LM11A-31 daily for three months before scanning the mice again and sacrificing them for autoradiography of brain tissue.
The APP mice took up much more of the tracer than the wild-type controls at baseline, indicating that neuroinflammation accompanies plaque pathology in these animals. Three months later, GE180 uptake in the cortex and hippocampus of animals treated with LM11A-31 had returned to wild-type levels, while untreated mice took up as much of the tracer as before. Analysis of the brain tissue showed lower levels of the glial activation markers Iba1 and TSPO in treated animals than controls.
James said LM11A-31 attenuates hyperphosphorylation and misfolding of tau, and prevents degeneration of neurites and synaptic spines. The compound seems to reduce inflammation as well, though it is unclear if this is a direct effect on microglia. Interestingly, James said she saw high GE180 uptake in the choroid plexus, where it also returns to baseline with treatment. The choroid plexus helps generate the cerebrospinal fluid and may serve as a conduit for removal of infiltrating immune cells.
William Trigg, a project leader at GE, told Alzforum that GE180 (which also goes by GEH120714 and flutriciclamide) is being tested in the clinic. General Electric has sponsored a Phase 1 trial for relapsing/remitting multiple sclerosis patients and healthy volunteers in Canada. Researchers at Imperial College in London have just kicked off investigator-sponsored trials of AD, as well as traumatic brain injury and multiple sclerosis. "We should see first data from those trials in 2015," said Trigg. The AD trial will enroll 30 people with AD, mild cognitive impairment, or normal cognition. Participants will also be scanned for brain amyloid.
Some people with a genetic variant in the TSPO receptor will be ineligible for GE180 imaging because their mitochondrial transporter binds poorly to the ligand. Trigg said that about 10 percent of Caucasian and African populations, and a smaller percentage in Asian populations, are homozygous for this variant, rendering them unsuitable candidates for the tracer. GE has implemented a blood test for binding affinity, and a genotype assay is available as well. Binding heterogeneity in the population could be a concern; however, Lemere said she does not expect the low-binding issue to hamper development of the tracer. Trigg encouraged researchers who want to plan studies using the compound to contact GE. "We have fairly open access," he claimed.—Tom Fagan
Move over, synaptic vesicles. Scientists are starting to appreciate that cells of the nervous system transmit information in a different type of vessel, as well. At the Society for Neuroscience annual meeting, held November 15 to 19 in Washington, D.C., researchers discussed the latest on exosomes—tiny bundles of cellular material that pass among cells. It appears that neuroglial cells in the brain use them like care packages. Astrocytes pack exosomes with specific messenger RNAs and then dispatch them to neurons, which soak them up and express the transcripts. However, some of the packages are bad news. Gioblastomas use exosomes to suppress the brain’s immune system, allowing the tumor to run rife.
“The neuroscience community is just catching on to this relatively novel form of communication," said Eva-Maria Krämer-Albers, Johannes Gutenberg University in Mainz, Germany. "Exosomes will completely change our view of how cells in the nervous system interact with each other,” she predicted. This basic biology could turn out to be important in Alzheimer's and other neurodegenerative diseases, as evidence has emerged that exosomes can transport and may even make toxic peptides such as Aβ and α-synuclein. (See Part 6 of this series.)
Exosome Primer—Vesicles Within Vesicles
All cells of the nervous system secrete exosomes. These small, 50-100 nm vesicles form when the membrane of a late endosome invaginates, then pinches off to make a small vesicle within the lumen of the endosome. This can occur many times, turning the endosome into a multivesicular body. These merge with lysosomes, spilling the vesicles into an acidic environment, which degrades them. However, if a multivesicular body instead fuses with the plasma membrane, the intraluminal vesicles escape into the extracellular space as exosomes (see image below). Exosomes may contain any of the molecules found in endosomes, including proteins and lipids, even RNAs specific to the cell that released them. Exosomes rid the cell of debris, but not all their cargo goes to waste. With the discovery that exosomes contain mRNA (see Valadi et al., 2007), scientists realized that these vesicles also could transfer information between cells in the nervous system.
Enter the Exosome: Multivesicular bodies (pink) that merge with the cell’s plasma membrane squirt exosomes (small gray-green circles), which may release toxic proteins such as Aβ and α-synuclein into the extracellular environment. [Image courtesy of Aguzzi and Rajendran, 2009, Neuron.]
A Regenerative Boost to Peripheral Neurons …
Researchers are trying to figure out which of the components exosomes carry are key for their communication. Felipe Court, Pontifica Universidad Católica, Santiago, Chile, studies how exosomes convey messages from Schwann cells to axons. These glial cells cozy up to mature axons in the peripheral nervous system and wrap myelin sheaths around them. Schwann cells inhibit the further growth of axons unless axons sustain damage, in which case Schwann cells undergo a sort of reprogramming. They then release exosomes that stimulate axonal regeneration when taken up by nearby sensory neurons in vitro, (see Lopez-Verilli et al., 2013). Just what in those exosomes might trigger the axons? Court suggested it was RNA.
Knowing that exosomes contain messenger RNA, scientists led by Court used high-throughput sequencing to identify what kinds of transcripts might be in exosomes from Schwann cells. The exosomes were rich in mRNAs that encode proteins involved in the assembly, organization, and regeneration of cells, as well as in axon growth and regeneration. This mRNA profile looked completely different from that of the original Schwann cells. “That’s very interesting because it means that the cell isn’t just putting RNA randomly into the vesicles, but is packaging specific transcripts for export,” Court said. Schwann cells concentrated the mRNA for neurofilament H, for instance, in their exosomes. Schwann cells do not actually make this protein. “This means that the cells are making transcripts, putting them in exosomes, and exporting them just for neurons to use,” said Court.
The exosomes also contained specific micro RNAs (miRNAs) that seemed to lend axons a helping hand. Out of 200 genes Court and colleagues identified as being involved in axonal regeneration and growth, in-silico analysis flagged 120 of them as targets of the particular miRNAs found in Schwann cell exosomes.
To find out how these exosomes affect axons, the group performed tests on cultured sensory neurons from the rat dorsal root ganglion. Isolating these neurons damages their axons, which begin to regenerate two to three days later. However, if the researchers added exosomes from Schwann cells to the sensory neurons, the axons began to regenerate immediately. The effect seemed unique to Schwann cell exosomes, as those from astrocytes did nothing to enhance axon growth.
Looking at the sensory neurons just a day after culturing, the researchers found a unique miRNA profile in the neurons treated with exosomes. Many miRNAs that predominated in the untreated neurons were absent in the treated ones. In fact, the miRNA profile of the latter resembled that of sensory neurons that had been damaged during isolation and were actively regenerating in culture. “Schwann cell exosomes are activating a specific set of genes [in the neurons],” said Court. In the future, researchers may be able to use exosomes to reveal new genes involved in axon regeneration, he said.
… and to Neurons in the Brain
In the same way that Schwann cells deliver exosomes to aid neurons in the periphery, recent studies suggest that oligodendrocytes, their central nervous system equivalents, shower exosomes onto nearby neurons. Krämer-Albers previously discovered that mouse oligodendrocytes secrete exosomes in response to synaptic activity in adjacent neurons (see Frühbeis et al., 2013). At SfN, she reported that in vitro, mouse cortical neurons pretreated with exosomes from oligodendrocytes turned on cell-survival pathways and resisted nutrient deprivation, oxidative stress, and ischemia. They also fired more often (see Fröhlich et al., 2014). The exosomes did not affect axon growth, however, nor did they protect neurons if they were applied after the stressor. “Unlike exosomes from Schwann cells, these don't seem to have regenerative potential, but rather, are neuroprotective,” Krämer-Albers said.
That's not to say that oligodendrocyte exosomes don't support axons. The German scientists found that, in mouse hippocampal neurons, the exosomes sped up anterograde transport of cargo down axons and cut down on the number of traffic stoppages. This suggests that the oligodendrocytes run a kind of “delivery” service, whereby neurons call on the helper cells to release neuroprotective care packages. “This may be a mechanism to maintain the long-term integrity of axons, which stretch far away from the nourishing cell body,” said Krämer-Albers.
Despite the growth of in vitro data on exosomes, scientists have yet to demonstrate that they work the same way in vivo. “It will be extraordinarily challenging to validate these findings in the human brain,” David Brody, Washington University in St. Louis, wrote to Alzforum. Krämer-Albers agreed, noting that the vast majority of exosome studies have relied on cultured cells because scientists lack the tools to manipulate exosome release in animals. She said the physiological role of exosomes may remain in question until the field comes up with specific ways to target their secretion and transfer in vivo.
Some researchers have come close. In a symposium, Vivian Budnik, University of Massachusetts Medical School, Worcester, reported that exosomes signal between pre- and postsynaptic cells at the neuromuscular junction in the fly brain (see Korkut et al., 2013). Those signals are crucial to maintain synaptic plasticity. She also found that Wnt signals important for fly brain development pass between neurons via exosomes (see Koles et al., 2012). “Though exosome biology is likely conserved through evolution, we still lack evidence for its function in the mammalian brain,” Krämer-Albers told Alzforum.
The Dark Side of Exosomes
Most of these findings put a positive spin on exosome signaling, but exosomes may not always be good for the brain. Tumors seem to exploit them to fuel their growth. One lethal brain tumor, glioblastoma multiforme, arises from astrocytes or their precursors. “We don’t know how these tumors are so aggressive, but we think the vesicles they release are priming the environment around them to allow them to progress more rapidly,” said Xandra Breakefield, Massachusetts General Hospital, Charlestown. These extracellular vesicles include exosomes and microvesicles released directly from the cell membrane (see image above). Researchers know that they help tumors develop new blood vessels and suppress immune cells that might rein in the cancers (see Iero et al., 2008 ). The RNA profile inside these vesicles differs starkly from that of the original tumor, and the cells that take up the vesicles translate the RNAs within them (see Skog et al., 2008).
Recently, researchers at Breakefield’s lab found that micro RNAs miR-451 and miR-21 are enriched in extracellular vesicles from glioblastoma multiforme cells and modify nearby microglia. While cultured microglia usually contain small amounts of these miRNAs, the cells rapidly took up gliobastoma exosomes, raising miR-451 and miR-21 levels 10- to 40-fold. Both these miRNAs target the messenger RNA for the transcription factor c-Myc. C-Myc expression fell in microglial cultures exposed to tumor vesicles. Breakefield is unsure how downregulation of c-Myc would modify tumor growth, but suggested that it may subdue the immune system, giving tumor cells free rein to grow. In a mouse glioblastoma model, microglia appeared to be attracted to the tumors. They took up tumor-derived vesicles, bumping up miR-451 and miR-21, while c-Myc expression fell.
Overall, the picture emerging is that extracellular vesicles transfer information through gene expression and induction of new properties in nearby cells, said Lawrence Rajendran, University of Zurich, who co-chaired the session. It remains to be seen how the RNAs are targeted to vesicles, how cells take them up, and how molecules get out of the exosomes once they enter the new cell, he added. “On top of that, the roles exosomes play in physiology and pathology are still to be determined,” he said.
Other researchers who heard the talks were intrigued. “The data are very convincing that exosomes are not just a way for the cell to dispose of things, but also to pass messages to other cells,” said Dimitrios Kapogiannis, National Institute on Aging, Baltimore. “I predict that exosomes will be an even hotter topic in the near future because more and more people are researching their biological function.” Scientists were particularly excited by the possibility that exosomes may propagate, and even generate, pathogenic proteins involved in neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases (see Part 6 of this series).—Gwyneth Dickey Zakaib.
Exosomes have emerged as players in cell-to-cell communication in the nervous system. At the Society for Neuroscience annual meeting held in Washington, D.C., November 15 to 19, scientists reported that these tiny, cell-derived vesicles may also contribute to pathology in neurodegenerative disease. Exosomes could make Aβ and carry it, and other pathogenic proteins such as tau, from one neuron to another, the story goes. At the same time, blocking the release of exosomes may lead to an accumulation of toxic proteins inside neurons, contributing to pathology.
Aβ-Generating Machines?
Exosomes are tiny packets of proteins, lipids, and nucleic acids that are released from most cells in the body. They form when the endosomal membrane folds in on itself and pinches off to create a vesicle inside the endosome’s lumen. Dozens of them jostle in endosome-derived multivesicular bodies. When those bodies fuse with the plasma membrane, the vesicles flood the extracellular space as exosomes (for details, see Part 5 of this series).
At SfN, Lawrence Rajendran, University of Zurich, explained that exosomes in neurons convene all of the working parts needed to make the Aβ peptide. He previously found that exosomes from neuroblastoma cells contained the β C-terminal fragment (CTF) of the amyloid precursor protein (APP) and Aβ. Others had reported that exosomes also contained β-secretase, but whether it was active was unclear (see Sharples et al., 2008). Could β- and γ-secretases possibly produce Aβ outside the cell? Studying exosome fractions from mouse primary neurons, Rajendran and colleagues have so far found BACE1 and all four components of the γ-secretase complex.
Exosomes and Parkinson's.
Exosomes accumulate as small vesicles in multivesicular bodies. Genetic variants in the PARK9 gene that cause an early form of Parkinson's limit their production (see text below).
[Image courtesy of Tsunemi et al., The Journal of Neuroscience, 2014.]
Though preliminary, their evidence suggests that the γ-secretase is active, because when incubated with APP β-CTF, the exosomes produced Aβ. No Aβ formed in the presence of a γ-secretase inhibitor, or when the exosomes came from mice lacking presenilins. This hints that exosomes carry the machinery for making Aβ. “We don’t know whether this is physiologically relevant, but it is an interesting observation,” Rajendran said. He emphasized that there is no evidence that exosomes make Aβ outside the cell in vivo. However, since amyloid-forming proteins are more prone to aggregation when they associate with membranes (see Butterfield and Lashuel, 2010), any exosome-associated Aβ could seed the aggregation of the peptide outside the cell, he said.
Do Exosomes ‘Pass It On’?
If exosomes offer cells a way to get rid of Aβ, then they could spread the peptide throughout the brain. For now, there is no evidence that neurons take up vesicles with pathogenic proteins in them, said Rajendran. Martin Hallbeck, Linköping University, Sweden, is studying this question in neurons derived from induced pluripotent stem cells made from healthy human fibroblasts. He incubated these neuron cultures with exosomes from neuroblastoma cells laden with fluorescently labeled α-synuclein or Aβ oligomers. Using confocal microscopy, he saw neurons ingest the exosomes and their proteins. When he then co-cultured these cells with fresh neurons that had not been incubated with exosomes, he saw that labeled oligomers of both α-synuclein and Aβ passed between them via exosomes. However, if he blocked endocytosis, the cells took up almost none. “These results suggest that exosomes provide at least one way for toxic aggregates to spread from neuron to neuron,” Hallbeck told Alzforum. While he and his research group had previously shown that oligomers transferred directly between neurons were toxic, he did not examine whether the exosome-delivered oligomers were, as well.
Neurons may not be the only ones propagating neurodegenerative proteins via exosomes. Tsuneya Ikezu, Boston University School of Medicine, believes microglia may do it, too. He thinks that microglia take up pathogenic aggregates from outside the cell and then spit some back out in exosomes, perhaps because they are hard to break down. To test this idea, Ikezu’s team injected the medial entorhinal cortex of wild-type mice with a virus encoding human tau. A special reporter that labeled virus-infected cells green distinguished them from the cells that might take up just transgenic tau. The human tau spread to the dentate gyrus over 28 days. Before it accumulated there, however, Ikezu noted activated microglia in the region, suggesting to him that neurons were sending advance warning along axons that terminate in the dentate gyrus, recruiting microglia. He thinks microglia took up the human tau and released what they were unable to digest in exosomes to be passed to other cells. In this experiment, preventing microglial activation greatly reduced tau spread in the mice. So did inhibiting neutral sphingomyelinase-2, which is required to make exosomes. Immunoelectron microscopy also revealed human tau inside the exosomes from the mouse brain.
This talk received mixed reviews. Dave Morgan, University of South Florida, Tampa, found it fascinating that reducing microglial activation slowed tau propagation. “Of course, there are many other reasons why microglial depletion could reduce tau spread, but this [elimination of microglial exosomes] is looking like a potential candidate mechanism,” he told Alzforum. Greg Cole, University of California, Los Angeles, also thought the findings were interesting. “This is a very new idea and the data to support it are novel and intriguing,” he wrote. However, he wondered if inhibiting neutral sphingomyelinase-2 could have similar effects on tau spread between neurons in the absence of a microglial effect. Cole noted that reducing microglial activation could reduce tauopathy independently of exosomes, for example, by diminishing tau phosphorylation.
Too Many Exosomes Are Bad, Too Few Even Worse
Rocío Pérez-González and Sebastien A. Gauthier, working in the lab of Efrat Levy at the Nathan S. Kline Institute, Orangeburg, New York, showed that brain cells from the postmortem tissue of people with Down’s syndrome expelled more exosomes than cells from people who had been healthy. Each vesicle also contained more APP and APP-CTFs. The researchers reported that cells in the Ts2 mouse model of Down’s do the same. Pérez-González suspects that a backup in the endosomal pathway forces the cell to release exosomes as a protective mechanism. “If the exosomes are enriched with toxic material, they may also be pathogenic and could help spread disease all over the brain,” she told Alzforum. The finding is in line with other research suggesting that diseased cells produce more exosomes, said Francesca Properzi, Superior Institute for Health, Rome. While the exosomes could result from a backed-up endosomal system, they could also simply serve as a way for cell to rid themselves of toxins.
Secreting too few exosomes could also pose a problem. Researchers led by Dimitri Krainc, Northwestern University, Chicago, have reported that α-synuclein accumulates inside fibroblasts from people with Kufor–Rakeb syndrome, an early onset form of Parkinson’s disease. Their fibroblasts make fewer intraluminal vesicles and release fewer exosomes than do cells from controls. Kufor-Rakeb results from a mutation in the PARK9 gene, a member of a family of ATPases that transport ions across membranes. Krainc found that overexpressing the PARK9 protein in mouse primary cortical neurons had the opposite effect, causing the cells to release synuclein into the medium. The researchers think PARK9 interacts with proteins of the endosomal sorting complex, which is required for making exosomes. They published this work in the November 12 Journal of Neuroscience (see Tsunemi et al., 2014). PARK9 defects were previously linked to lysosomal dysfunction (see Jun 2012 news story on Dehay et al., 2012).
Given that exosomes appear to be mixed up in neurodegenerative disease, might they offer a therapeutic target? Since exosomes appear to rid cells of waste, interfering with their entry into recipient cells might be better than inhibiting their release. Xandra Breakefield, Massachusetts General Hospital, Charlestown, who co-chaired the session with Rajendran, said that if specific interactions mediate exosome uptake into particular cells, those interactions could be selectively blocked. Very little is known about uptake mechanism at this point. Some groups are working to develop exosomes as biomarkers of neurodegenerative diseases, including AD and PD (see Aug 2014 news story; and Shi et al., 2014), recommended that more basic biology be studied before trying to target these vesicles.—Gwyneth Dickey Zakaib
Most amyloid fibrils in the human body are toxic, but a select few perform important functions. What sets them apart? At the Society for Neuroscience annual meeting, held November 15 to 19 in Washington, D.C., Guillaume van Niel from the Institut Curie in Paris suggested that it could be ApoE. The lipoprotein concentrates on vesicles where fragments of the premelanosome protein (PMEL) congregate, and seems key to their orderly fibrillization. PMEL amyloid serves as a scaffold for the pigment melanin. Van Niel said the findings may help scientists understand how Aβ interacts with ApoE4, the strongest genetic risk factor for Alzheimer’s disease.
Pigment cells create PMEL fragments in a way that parallels processing of the amyloid precursor protein (APP) (see Nov 2008 news story). PMEL traffics from the Golgi to the plasma membrane and then to endosomes. There, BACE2 sheds the large extracellular domain of PMEL into the lumen of the endosome, while γ-secretase further processes what's left (see image below). In the case of PMEL the shed portion (the equivalent of sAPPβ) goes on to be cleaved by other proteins, yielding fragments that make amyloid fibrils on the intraluminal vesicles. The amyloid scaffolds are then shunted off to melanosomes, where they concentrate melanin.
Parallel Processing? PMEL follows a similar trafficking and processing path to APP, and forms fibrils on intraluminal vesicles inside multivesicular bodies. [Image courtesy of Guillaume van Niel in Rajendran et al., 2014 J Neurosci.]
To figure out what makes the PMEL fibril harmless, van Niel took a close look at where they form. He snap-froze exosomes—intraluminal vesicles that had been expelled from the cells (see Part 5 and Part 6 of this series). Viewed up-close with an electron microscope, he noticed a cap-like structure on the outer membrane of these vessels. The cap’s cylindrical structure comprised multiple layers separated by 3.5 nm each. After two years of trying to figure out what the cap was, van Niel spotted a similar structure in an electron microscope image of a lipoprotein particle—multiple layers separated by 3.5 nm. Using a type of mass spectroscopy, he and colleagues checked if exosome caps contained any apolipoproteins. There was indeed one enriched in exosomes sporting caps compared to those without—none other than ApoE. “We got excited because a variant of ApoE is the primary genetic risk factor for sporadic AD,” said van Niel. Given the parallels between APP and PMEL processing, this finding could offer a clue about how ApoE interacts with APP’s fragments, he said.
To see if the ApoE cap interacted with the amyloid fragment of PMEL, van Niel used co-immunoprecipitation and found that the two bound each other. Electron microscopy revealed that this happened on the surface of the intraluminal vesicles inside multivesicular bodies of pigment cells. If the scientists depleted ApoE mRNA from the cells, the PMEL did not fibrillize. Instead, it formed unstructured aggregates in the lumen of the multivesicular body. The same organization defects plagued pigment cells from mice genetically deficient in ApoE. This suggested that ApoE is necessary to form the PMEL fibril on the vesicle outer membrane.
Scientists at the conference found this discovery extremely interesting. “What makes it so striking is that the cell traffics and cleaves PMEL similarly to APP,” said Gunnar Gouras, Lund University, Sweden. He thought it might help researchers understand the relationship between Aβ and ApoE, saying “We have been slow to understand how ApoE ties into Alzheimer's." Others remained to be convinced. “I was intrigued by this presentation, though it is hard to understand how it could relate to Alzheimer’s disease,” said Martin Ingelsson, Uppsala University, Sweden.
Van Niel said his findings suggest ApoE has a natural affinity for amyloid-forming peptides, and tends to fibrillize them. Since neurons do not normally produce ApoE, it only encounters their Aβ peptides after they leave the cell, whereupon ApoE may nucleate formation of fibrils (see image below).
Put a Cap on It: In pigment cells (right), ApoE caps (green) nucleate ordered PMEL fibrils on intraluminal vesicles (yellow). In the brain (left), ApoE may encounter Aβ peptides (red) outside the cell. [Image courtesy of Guillaume van Niel.]
Gouras looked at this a slightly different way. He speculated that ApoE does make its way into neuronal endosomes in AD. Neurons might take up ApoE from glial cells, or they may produce it themselves after injury, he said (see Xu et al., 2006). If so, then ApoE and Aβ may interact inside the cells after all.
Ingelsson recommended testing exosomes from AD-related cell systems for ApoE. Lawrence Rajendran, University of Zurich, Switzerland, and van Niel together are looking for ApoE caps on exosomes from astrocytes. If there, they want to know if they help Aβ form fibrils, and whether caps made from the different isoforms of ApoE—ApoE2, or ApoE3, or ApoE4—do this differently.—Gwyneth Dickey Zakaib
The genes involved in Alzheimer’s disease harbor both pathogenic and protective factors, and at the annual meeting of the Society for Neuroscience, November 15 to 19 in Washington, D.C., researchers proposed two new variants that might have a hand in causing disease. Genotyping members of a family with a history of late-onset AD turned up the netrin receptor UNC5C. Targeted sequencing revealed the Tmp21 gene, a proposed modulator of γ-secretase. Other researchers reported that whole-exome sequencing of people homozygous for the ApoE4 allele uncovered variants that may ward off late-onset disease. And data gleaned from genetic studies have led yet others to design a therapeutic that promotes clearance of Aβ in the brain.
All in the Family: UNC5C
Late-onset AD runs rampant in some families, yet in many cases the responsible genetic risk factors remain hidden. Monica Wetzel–Smith of San Francisco-based Genentech explained how she and her colleagues looked for variants that associate with AD in one extended family with a pattern of late-onset disease that resembled autosomal dominance. The researchers sequenced the entire genome of one affected family member and the exome of an aunt, who was the most distantly affected relative. Both turned out to share the T835M missense variant in UNC5C. Six relatives with the disease, but also two unaffected ones, had one copy of this variant.
Four other families from the LOAD family cohort (see Wijsman et al., 2012) carried T835M UNC5C as well. In one, all four people with the disease did. One person in each of two other families carried the allele, but neither had AD. Because one of these carriers was younger than 70, the scientists are uncertain whether he or she will get the disease. In the fourth family, three people carried the variant. All three were cognitively healthy but younger than 70, so Wetzel-Smith said she could not infer whether the allele was involved in the family’s disease. To see if the variant is enriched in other people with late-onset Alzheimer’s, the researchers compared the genotypes of more than 8,000 AD patients with almost 100,000 controls. These came from the Genentech cohort, Washington University in St. Louis, the Alzheimer’s Disease Genetics Consortium at the University of Pennsylvania, Philadelphia, and the Iceland-based deCODE Genetics study. Seventeen patients (0.2 percent) and 68 controls (0.07 percent) carried the mutation. The researchers concluded that the T835M variant doubled the odds of getting Alzheimer’s.
What does this variant do? UNC5C is an axon guidance receptor for netrins, which are proteins secreted into the cellular environment during brain development. The receptor is thought to influence apoptotic signaling, much like its cousin UNC5B (see Wang et al., 2009). Both receptors contain a cytosolic “death domain.” It remains buried inside the protein structure when a netrin is bound. In the absence of ligand, the protein undergoes a conformational change exposing the death domain; this recruits signaling molecules that relay apoptotic messages to the nucleus. T835M seems to alter the “hinge” region of the protein's intracellular domain, to keep the death domains constantly exposed. In culture, fewer HEK293T cells survived transfection with T835M UNC5C than with wild-type protein. Aβ and tau production were unaffected. Rat hippocampal neurons expressing the risk variant succumbed more quickly to Aβ-, glutamate-, or staurosporine-induced toxicity.
The researchers published the findings in the November 24 Nature Medicine, where they suggested that T835M UNC5C predisposes carriers to neurodegenerative diseases (see Wetzel et al., 2014). Robert Graham co-led the work with Ryan Watts at Genentech in San Francisco and Carlos Cruchaga and Alison Goate at Washington University in St. Louis. Graham told Alzforum that carrying the UNC5C variant may make neurons more susceptible to stress, exacerbating any underlying neurodegenerative processes. The authors found this protein highly expressed in neurons of people’s temporal lobe and hippocampus.
“While it is a rare variant, it clearly looks like the UNC5C mutation increases AD risk,” David Holtzman, WashU, wrote to Alzforum. He was particularly interested that the mechanism seemed unrelated to Aβ. “The data looked good,” agreed Steve Estus, University of Kentucky, Lexington. “This is another indication that Alzheimer’s is a multiple etiology disease," he said. "I wouldn’t have thought that a cell death pathway would be implicated in Alzheimer’s, but that’s how it looks.” Other researchers are skeptical. John Hardy, University College London, said he would reserve judgment until the findings are repeated. “Rare variant work is extremely difficult to prove, and just as difficult to refute.”
Zeroing In on Suspects
Other researchers are taking a targeted approach to finding risk alleles, looking in suspect genes. For example, the type I transmembrane protein TMP21 has been reported to modulate γ-secretase cleavage of APP (see Chen et al., 2006, and Vetrivel et al., 2007), but no one had found a genetic link between this receptor and Alzheimer’s.
Xiaojie Zhang, a Ph.D. student in the lab of Weihong Song, University of British Columbia, Vancouver, Canada, pored over intron and exon sequencing data from 261 AD patients and 236 controls. She found a single nucleotide polymorphism (SNP) in intron 4 of TMP21 that was associated with a 1.5 odds ratio for AD. The scientists have not looked for this association in any other cohorts yet, and Song would not say how many of the patients and controls carried the risk allele.
In HEK293 cells, the variant accelerated maturation of Tmp21 pre-mRNA to mRNA, meaning the SNP made splicing more efficient. Cells harboring the SNP made more Tmp21 protein. To see whether this SNP affected Aβ production, the researchers transfected HEK293 cells expressing APPSwe with plasmids encoding either wild-type Tmp21 or the variant. Production of both Aβ40 and 42 crept higher in the latter. Zhang suggested that perturbing TMP21 could affect the risk for Alzheimer’s disease. Holtzman wondered if Aβ40 or 42 levels differed among people with the variant and those without. Zhang said she had not looked at that yet.
“Reproducing this genetic association in larger cohorts is critical,” said Estus. “In the past, many candidate gene studies identified genetic risk factors in cohorts of this size that were not reproduced in other populations.”
Help Wanted: Seeking the Protectors
What about genetic variants that ward off disease? Researchers are studying those who are most at risk for developing Alzheimer’s, but still cognitively normal, to find variants that might delay AD. Aurelie N’Songo, of the Mayo Clinic in Jacksonville, Florida, who works in the labs of Nilüfer Ertekin-Taner and Guojun Bu, has examined the genetic makeup of people who are homozygous for the ApoE4 allele, older than 75, and cognitively intact. People with two copies of this allele have up to a 15-fold greater chance of developing late-onset Alzheimer’s disease by age 75. Only about 2 percent of the population is ApoE4 homozygote and those who are also older than 75 and have no cognitive impairment are even harder to find. Trolling the collection of the Mayo Clinics in Rochester, Minnesota, and Phoenix, N'Songo found 24 European-Americans who were ApoE4/4 with an average age of 81.
Sequencing the entire exome of these people, N'Songo found more than 200 variants enriched in this group compared with the general population (see NHLBI GO Exome Sequencing Project). Exome data from the Mayo Clinic Biobank of 88 people with mixed ApoE genotypes indicated 40 of those variants were specifically enriched in non-demented ApoE4 homozygotes. The preliminary data has not been validated yet, Ertekin-Taner emphasized. That said, it places some of these variants in genes previously associated with AD risk, such as HLA, ADAM9, ADAM29, and GSTM1. Other variants fit into pathways that may be associated with Alzheimer’s, such as cell signaling and oxidative stress.
Has the group checked ApoE4 homozygous people with dementia to see if they lack any of the 40 variants? N’Songo said they are trying to answer that question with ADNI data. They also plan to analyze data from 11,000 people in the AD Sequencing Project to see if those variants are enriched in non-demented ApoE4/4 carriers. Giulio Taglialatela, University of Texas Medical Branch at Galveston, asked if any of these 24 homozygous individuals had AD pathology in the brain. N’Songo said her group lacks histopathological information on these people, because they are still alive and only gave blood. “I am impressed by the innovative strategy they are using,” Taglialatela told Alzforum.
Capitalizing on the Data
Typically, genetic data do not lead directly to therapy development, but consider this project: Manasi Malik, from Estus' lab, is working on inhibiting the transmembrane receptor CD33, a known AD risk factor (see Aug 2013 news story). CD33 is present on immune cells in the periphery and the brain. This receptor prevents microglia from ingesting Aβ (see Gricuic et al., 2013). A protective, non-functional variant removes that brake and allows the cells to gobble up more Aβ. Malik tests whether antibodies to CD33 might do the same. One such antibody, Lintuzumab, has been used, to no avail, in people with leukemia, where CD33 is overexpressed.
Malik reported that 10 ng/ml of Lintuzumab reduced CD33 on the microglia cell surface by 80 percent. Estus said they are now testing whether this treatment improves microglial phagocytosis of amyloid and dead neurons, and affects cytokines released by microglia. He is unsure how much Lintuzumab crosses the blood-brain barrier, but thinks it may still be a viable therapeutic option even if it only acts in the periphery because peripheral monocytes cross into the brain to clear up Aβ deposits (see Dec 2014 news story).—Gwyneth Dickey Zakaib
Prion protein, insulin receptors, immunoglobulin-like receptors, PirB, unspecified membrane phospholipids, the list goes on … Now, add another purported Aβ receptor. At the Society for Neuroscience annual meeting in Washington, D.C., November 15 to 19, researchers led by Susan Catalano at the Pittsburgh-based biotech Cognition Therapeutics (CogRx) reported that Aβ interacts with the sigma2 receptor, which also goes by the alias progesterone receptor membrane component 1. Small molecule ligands developed by the company bind sigma2/PGRMC1 and reportedly restore cognitive function in transgenic mouse models of AD. The ligands block all manner of Aβ species from accessing the receptor. They even displace Aβ oligomers from brain tissue of Alzheimer’s disease patients, Catalano claimed. Could sigma2 be the real McCoy? "What is different is that Aβ no longer binds when the researchers knocked down sigma2," said John Cirrito, Washington University, St. Louis. "The pharmacology was interesting, but the genetics was the clincher for me," he said. Cirrito consults for the company.
Aβ Receptor?
Primary hippocampal neurons express sigma2/PGRMC1 (red), which often co-localizes with synaptophysin (green) in synaptic boutons. [Image courtesy of Izzo et al., PLoS One.]
Iryna Benilova from K.U. Leuven, Belgium, wrote to Alzforum, "This study is outstanding in three main aspects: compound-mediated displacement of bound oligomers has not been shown for other Aβ receptors; the neutralizing effect of sigma-2 allosteric antagonists holds for a remarkably broad array of Aβ species; and finally, the in vivo effects of sigma-2 antagonists on synapses and memory in two AD mouse models point to an interesting therapeutic potential of these compounds" (see full comment below).
Despite its name, sigma2/PGRMC1 does not bind progesterone. Little is known about the protein, though it seems to stabilize a variety of other receptors on the cell membrane. Catalano and colleagues identified sigma2 after running an unbiased screen for chemicals that prevent Aβ from interfering with the transport of vesicles to the cell surface. At the SfN meeting in San Diego in 2010, Catalano introduced CT0093. Found in a proprietary chemical library, this molecule restored vesicle trafficking in primary hippocampal neurons treated with Aβ oligomers from human tissue (see Dec 2010 conference story). Since then, the company scientists have identified other compounds with similar properties, and those eventually led them to the sigma2 receptor. They described their work on five posters at this year's meeting and in two recent back-to-back papers in PLoS One (see Izzo et al., 2014, and Izzo et al., 2014).
Four of the CogRx compounds—CT0109, CT0093, CT01344, and CT01346 (structures can be found in the PLoS papers)—almost completely prevented a variety of Aβ species (including synthetic oligomers) and patient-derived oligomers from binding to primary hippocampal neurons. In her poster, Kelsie Mozzoni reported that the compounds protected vesicle trafficking when added before Aβ oligomers and restored membrane trafficking when added after. They also prevented loss of synaptic spines. In vivo, the compounds readily entered the mouse brain when given subcutaneously (CT0109, CT0093) or by mouth (CT01344, CT01346). They protected animals from Aβ-associated memory loss in acute and chronic dosing regimens. These included injecting the compounds into the hippocampi of normal mice an hour before injecting Aβ oligomers into the same location, giving the compound by mouth to 11-month-old transgenic APP mice for 42 days, and a 5½-month treatment begun when APP mice were 9 months old.
To find out how these molecules work, the researchers screened 100 known cell surface receptors and proteins. The compounds bound specifically only to sigma2/PGRMC1. Interestingly, they only restored memory in the APP mice when their concentrations in the brain were sufficient to occupy 80 percent of sigma2 binding sites. Fifty percent occupancy had no behavioral effect. "That makes us believe sigma2 is the true target [for these compounds] in vivo," said Catalano.
Knocking down sigma2 with siRNA eliminates Aβ binding sites. [Image courtesy of Izzo et al., PLoS One.]
Other experiments suggest these compounds are antagonists of Aβ. In her poster, Courtney Rehak showed that if she silenced the receptor in primary rat neuronal cultures using siRNA, Aβ oligomer binding plummeted more than 90 percent. Colleen Silky's work demonstrated that the CogRx compounds not only competed with Aβ for binding to primary hippocampal neurons, but displaced the peptide when they were added later. "I am not aware of any small molecule that can both prevent Aβ oligomers from binding and displace those oligomers," said Catalano. "That is a major difference with our work."
The observations extend to human tissue. Collaborating with Tara Spires-Jones, University of Edinburgh, the CogRx researchers found that CT1344 displaced endogenous Aβ from postmortem brain slices. Nicholas Izzo incubated tissue from six AD patients for one hour with either the compound or antibodies to sigma2, and saw a dose-dependent loss of Aβ immunoreactivity. In the AD brain, sigma2 expression may be dysregulated, as well. Izzo and colleagues found that brain tissue from patients with advanced dementia expressed as much of the protein as did tissue from age-matched controls, while levels of another receptor (Sigma1) had dropped by about half. The authors argue that because people with advanced dementia have lost neurons, sigma2 must be upregulated in their brain for it to match control levels.
How might sigma2 mediate Aβ toxicity? Catalano acknowledged that it is not clear if Aβ binds to sigma2, or to something else that interacts with sigma2. "Both are possibilities," she said, "The story is more subtle than just another Aβ receptor."
Since sigma2 appears to be involved in trafficking receptors on the cell surface, might it interact with some of the previously identified Aβ partners? "That's absolutely the first place you might start to look to understand the oligomer receptor complex," said Catalano. Carla Shatz, Stanford University, California, agreed. "It will be fascinating to see how their discovery meshes with our finding that PirB is a high-affinity receptor for Aβ," she wrote to Alzforum (see Sep 2013 news story).
CogRx hopes to start clinical trials next year. "In light of the recent clinical trial failures, both of our studies offer hope that there will be new (earlier) treatment avenues that do not depend on busting up plaques at times when it may be too late to have any effect," said Shatz.—Tom Fagan
In mouse models of Alzheimer's disease, before cognition fails, tau fibrillizes, or Aβ even begins to accumulate, calcium signaling goes awry in the brain as neurons release more of the cation from their intracellular stores than they should. At the Society for Neuroscience annual meeting, held November 15 to 19 in Washington, D.C., Grace (Beth) Stutzmann, Rosalind Franklin University, North Chicago, Illinois, revealed new data to explain how this calcium overload damages dendritic spines, diminishes synapses, and dampens their ability to release vesicles. Stutzman claimed that this imbalance impairs synaptic plasticity not only in single cells, but in the entire hippocampal network. She attributed the effects to excessive release of calcium through the ryandodine receptor (RyR) calcium channel. Stutzmann is developing drugs to restore normal ryanodine (RyR) receptor function, and introduced several compounds at the meeting.
The RyR receptor is a large, calcium-activated calcium channel that sits in the membrane of the endoplasmic reticulum (ER). People have three different isoforms. RyR1 predominates in skeletal muscle, while RyR2 and RyR3 are found in cardiac cells and neurons. Though inside cells, these receptors are sensitive to neurotransmission. During an action potential, calcium enters through ligand-gated channels on the plasma membrane and opens RyR receptors on the ER. The ER then releases some of its stored calcium through the RyR. Researchers, including Stutzmann, reported that RyR2 expression rises in AD mouse models and in people with the Alzheimer's (see Stutzmann et al., 2006; and Zhang et al., 2010). In 3xTg-AD mice, synaptic stimulation caused more calcium to flow through the RyR than in control mice (see image below) (see Aug 2009 news story on Chakroborty et al., 2009). In turn, the neurons were susceptible to synaptic depression (see Chakroborty et al., 2012). Stutzmann now finds that synapses begin to wither under this elevated calcium signaling, wearing down their ability to function.
Overreaction:
In response to a synaptic stimulus, the dendrite of a 3xTg-AD mouse (right) releases three times as much calcium as a control dendrite. [Image courtesy of Grace (Beth) Stutzmann.]
Researchers in Stutzmann's lab compared 3- and 4-month-old wild-type to age-matched 3xTg-AD mice, before they developed AD-related pathology or cognitive deficits. Calcium irregularities had already begun (see Stutzmann et al., 2006). Using electron and confocal microscopy to examine synapses between the CA3 and CA1 area of the hippocampus, the researchers found that CA1 dendrites sported fewer mushroom spines, a type important for encoding long-term memories. CA1 and CA3 neurons formed fewer intact synapses with each other. The protein-rich region called the postsynaptic density, which is responsible for interaction with the presynaptic cell, became smaller. Presynaptically, the pool of neurotransmitter vesicles poised for release was drained.
The group found that last result particularly interesting. Could the depleted vesicles explain the synaptic depression Stutzmann’s group had previously found in these neurons? In patch-clamp recordings, the group found that these CA3 neurons spontaneously released more neurotransmitter vesicles than wild-type CA3 neurons. Stutzmann believes this constant spitting out of vesicles depletes the store available for release when a stimulus arrives. Then, fewer vesicles are released, stimulating synaptic depression rather than potentiation.
Did all this happen because of the excess calcium from RyR2 receptors? To find out, the researchers treated 3xTg-AD mice with dantrolene, an allosteric modulator that dampens RyR activity. The compound normalized calcium signaling in 3xTg-AD and TASTPM mice (see Chakroborty et al., 2012). At SfN, Stutzmann showed that dantrolene returned to normal the number of spines, synapses, and spontaneous vesicle released.
Dantrolene reduced tau and Aβ pathology in the PS/APP transgenic mice (see image below). Reducing calcium in the cells may dial back the activity of tau kinases and BACE1, Stutzmann said. “The extent of the structural and functional changes due to calcium dysregulation was profound,” she told Alzforum. “I was surprised that these were all reversed by stabilizing calcium signaling.” While she has no behavioral data on these mice, other groups have reported that dantrolene prevents and reverses cognitive deficits (see Peng et al., 2012, and Oulès et al., 2012).
A Reduced Load: After four weeks of dantrolene treatment (right) in PS/APP mice, Aβ pathology falls by half in the hippocampus and overlying cortex compared to saline-treated controls (left). [Image courtesy of Grace (Beth) Stutzmann.]
Gregory Brewer, University of California Irvine, noted that synapse loss accounts for the lion’s share of cognitive defects in the disease. “Any handle we can get on the reasons for synapse loss gets us closer to the cause of the disease, and is an obvious link to memory impairment,” he said. Did the changes in spines, synapses, and vesicles in the 3xTg–AD mice correlate with Aβ and tau pathology? Stutzman plans to address this question in the future.
Network Trouble
To address whether the synaptic deficits affect transmission across neural networks, Stutzmann, with William Frost and Shreaya Chakroborty, incubated hippocampal slices with a lipid-soluble fluorescent dye that absorbs more light and then fluoresces more intensely when the voltage across the cell membrane becomes more positive. A high-speed camera taking more than 1,000 pictures per second visualized the voltage changes through the hippocampal network. In control slices, a first synapse-strengthening stimulus causes a subsequent pulse to propagate farther through the network and depolarize neurons for longer than the first; both the greater distance and time are evidence of potentiation. However, in slices from 3xTg-AD mice, the second pulse spread no farther and lasted no longer than the first. This suggests, for the first time, that with elevated calcium release, the propagation of signals through the hippocampus is blunted in space and time. “It means regions of the hippocampus are less effective in transmitting information and sustaining it as they should be,” Stutzmann said. Next, she will test whether treatment with a RyR2 modulator restores network transmission.
“This is a great new method to image hippocampal networks,” said Olivier Thibault, University of Kentucky, Lexington, noting that it visualizes depolarization across large areas of the brain.
Looking for drug candidates that enter the brain better and are more specific than dantrolene, Stutzmann worked with medicinal chemists at her university, who synthesized allosteric RyR2 modulators. Stutzmann introduced four of the compounds, CK013, CK017, RD14, and RD95. All four normalized calcium responses in hippocampal slices from 3xTg mice. They did not markedly change other calcium-dependent properties of neurons, such as maintenance of membrane potentials. Given to 3-month-old 3xTg mice over four weeks, CK013 and CK017 restored RyR-induced calcium release to control levels. CK013 reduced tau and Aβ pathology in the hippocampus by more than half when given to 6-month-old TgCRND8 mice. Ongoing studies are testing whether this treatment preserves synaptic function and structure, and cognition in mice, Stutzmann said.
“These new derivatives of dantrolene that are effective in the central nervous system have provided new and exciting possibilities of treating patients before symptoms are prominent,” wrote Gary Gibson, Weill Cornell Medical College, New York, to Alzforum. “The remaining challenge is to show this approach is effective in human tissues and at non-toxic dosages.” Dantrolene itself is used as a muscle relaxant and approved for the treatment of malignant hyperthermia, an inherited disease of rising body temperature and muscle spasms.—Gwyneth Dickey Zakaib
Do Astrocyte Receptors Go Over the Top in Alzheimer’s?
Astrocytes perform an elaborate array of support functions for neurons, but if they get out of sorts they can lead to trouble. At the Society for Neuroscience 2014 Annual Meeting, held November 15 to 19 in Washington, D.C., researchers reported on two receptors that might pack a punch in astrocytic cell membranes in Alzheimer’s disease. Astrocytes near plaques overexpress the P2Y1 purinergic receptor, which appears to be involved in astrocyte hyperactivation. Meanwhile, the related adenosine A2A receptor may impair memory, as knocking it out improved the performance of both wild-type and AD transgenic mice in behavioral tasks.
Purinergic receptors sit on the plasma membrane and are activated by ATP or its breakdown products ADP, AMP, and adenosine. In addition to playing a role in cell metabolism, these molecules signal stress or “danger” among brain cells. The receptors comprise two main classes, P1 and P2. P1 are G-protein-coupled receptors activated by adenosine. They include the A2A subtype. P2 receptors are activated by ATP or ADP, and can be either ionotropic, in which case they are called P2X, or G-protein-coupled, called P2Y. The P2Y1 subtype signals the endoplasmic reticulum to release calcium. P2Y1 and A2A normally exist at low levels on astrocytes, but in neurodegenerative disease, they appear to run amok.
Unusual expression:
Reactive astrocytes (red) strongly express P2Y1 receptors (green, overlap in yellow) around Aβ plaques (blue).
[Image courtesy of Delekate et al., Nature Communications.]
Early in AD, astrocytes in the vicinity of amyloid plaques exhibit a higher degree of spontaneous calcium activation than usual (see Kuchibhotla et al., 2009). Andrea Delekate, working with Gabor Petzold and colleagues at the German Center for Neurodegenerative Diseases (DZNE) in Bonn, went searching for the cause of this hyperactivity. Delekate implanted a cranial window in anaesthetized APPPS1 mice and monitored calcium activity in astrocytes with multiphoton microscopy. She first confirmed that the astrocytes were hyperactive, then tested whether these star-shaped cells were simply responding to neuronal activity by blocking either neuronal transmission or the astrocyte metabotropic glutamate receptors that sense it. Neither calmed the astrocytes.
Next Delekate focused on ATP receptors. In response to inflammatory signals, astrocytes, and other cells release ATP to alert surrounding neurons and glia. Previous research suggests that cells differentially express these receptors in Alzheimer’s disease, depending on the receptor subtype (for a review, see Franke et al., 2012). Could one of them explain the calcium hyperactivity? Using specific inhibitors for each purinergic receptor subtype, Delekate found that blocking P2Y1 returned astrocyte signaling to normal in the APPPS1 mice. She also confirmed previous evidence that astrocytes express unusually high amounts of P2Y1 around plaques (see image above and Moore et al., 2000). In wild-type mice, the P2Y1 inhibitor left normal astrocyte function unchanged. Delekate reported these findings, with co-first author Martina Füchtemeier, in the November 19 Nature Communications (see Delekate et al., 2014).
The researchers are now testing whether infusing the P2Y1 inhibitor into the ventricles of the brain in APP/PS1 mice for several weeks returns astrocyte activity to normal. Greg Cole, University of California, Los Angeles, questioned whether reducing astrocyte hyperactivity would be useful, since the activated astrocytes around plaques may be there to remove Aβ. “We don’t know yet if it is a good or bad thing,” said Delekate. She will explore that in treatment studies targeting different age points and disease stages to see if blocking the receptor affects memory function and disease progression.
Purines Purge Memories? Anna Orr, who works in the lab of Lennart Mucke, Gladstone Institute of Neurological Disease, San Francisco, claimed that astrocytes in AD mouse models also overproduce the A2A receptor. Expressed by neurons and astroglia, the A2A receptor couples to Gs proteins that raise the levels of the second messenger cAMP in the cytosol. Previous studies suggested that A2A renders neurons more susceptibility to AD pathology, and that A2A antagonists prevented Aβ-induced cognitive deficits in mouse models of the disease (see Rahman, 2009, and Dall'Igna et al., 2007). However, scientists know little about how A2A expression changes in Alzheimer’s.
Orr and colleagues expected to find A2A in microglia, because they had previously found that these cells ramped up its expression upon inflammation (see Orr et al., 2009). To their surprise, they found that astrocytes, not microglia, tested positive for A2A in postmortem tissue from people with AD. The hippocampus expressed the receptor most strongly, and levels correlated positively with Braak staging—the more advanced the disease, the more A2A was expressed. Similarly, they found A2A to be up in several transgenic AD mouse lines, where the receptor accumulated once plaques started to form. The group wondered if this overexpression might damage learning and memory.
To find out, Orr and colleagues conditionally knocked out one or both copies of the gene for the A2A in the astrocytes of wild-type mice, leaving their neuronal A2A intact. These mice performed as well as controls when learning to navigate the Morris water maze, but better remembered the platform location several days later. In the same way, removing astrocytic A2A receptors in J20 mice that expressed human mutated APP improved their memory performance. Only old mice appeared to benefit from the lack of A2A—young knockouts performed no better than control wild-type mice.
In a different set of experiments, Orr elevated astrocyte Gs-coupled signaling by expressing a mutated serotonin receptor in the astrocytes of adult mice. The receptor has been engineered to bind only a synthetic ligand, and adding this agonist just before or after training the mice in the Morris water maze impaired their memories a few days later. “Together, the results imply that increases in astrocytic A2A with age are contributing to memory loss, and removing that A2A is beneficial,” Orr said.
The same could be true for people, she suggested. When given the A2A receptor blocker caffeine, people better consolidated long-term memories in a discrimination task (see Borota et al., 2014). Orr and others in the Mucke lab are now looking into what might cause A2A to be overexpressed, and what downstream mechanisms of the receptor cause these memory changes.
“The data are really interesting as they reinforce the idea that A2A receptor dysregulation may drive pathogenic mechanisms involved in AD,” David Blum, INSERM, Lille, France, wrote to Alzforum. He noted that previous studies have reported neuronal increases in A2A in the hippocampus of aging rats (see Canas et al., 2009). In addition, evidence suggests that blocking A2A protects synapses from Aβ (Canas et al., 2009) and tau (Laurent et al., 2014). “This work adds another piece to the A2A puzzle and argues for testing A2A receptor antagonists currently in trials for Parkinson’s disease in AD,” Blum wrote. A2A antagonists have shown some minor benefits for people with Parkinson’s disease as an adjunct therapy to dopaminergic drugs (see Jenner et al., 2014). One, istradefylline, has been extensively studied in Parkinson’s but was not approved by the FDA.—Gwyneth Dickey Zakaib
Laurent C, Burnouf S, Ferry B, Batalha VL, Coelho JE, Baqi Y, Malik E, Mariciniak E, Parrot S, Van der Jeugd A, Faivre E, Flaten V, Ledent C, D'Hooge R, Sergeant N, Hamdane M, Humez S, Müller CE, Lopes LV, Buée L, Blum D.
A2A adenosine receptor deletion is protective in a mouse model of Tauopathy.
Mol Psychiatry. 2014 Dec 2;
PubMed.
Nanobubbles: Potent Potion, or Just Effervescence?
The tinier the bubbles in a glass of champagne, the quicker the booze goes to your head, some say. At the Society for Neuroscience meeting held November 15 to 19 in Washington, D.C., scientists presented surprising findings that suggest the smallest bubbles of all—nanobubbles—may boost cognition and even fight Alzheimer’s disease. The bulk of the data came from Revalesio, a small company based in Tacoma, Washington, and its collaborators.
Nanobubbles are not found in Dom Perignon. In Revalesio’s formulation, they are charged microscopic bubbles filled with oxygen, not carbon dioxide. Researchers have reported that these tiny spheres achieve all manner of cellular heroics, including soothing neuroinflammation, blocking tau phosphorylation, saving neurons from the brink of death, boosting synaptic plasticity, and even rescuing memory deficits in AD mouse models. Clinical trials are already underway to treat multiple sclerosis and asthma, with one for AD on the horizon. Research into the bubbles and their potential applications has taken off recently: A conference devoted to nanobubbles took place in Shanghai last October.
Powerful Packages.
Charged ions line the surface of nanobubbles filled with oxygen in this depiction of the structure of RNS60. [Image courtesy of Roy et al., PLOS One, 2014.]
The way these wee bubbles work their magic remains a mystery, and many are still skeptical. However, researchers reported that most of their effects can be explained by activation of phosphatidylinositol-3-kinase (PI3K) and/or a boost in mitochondrial function.
Nanobubbles were discovered serendipitously in the late 1990s by Tony Wood, a then-retired Texas Instruments scientist. In his garage, he tinkered with ways to mix gases and liquids more efficiently. When his mixtures retained dissolved oxygen for longer than expected, Wood discovered that nanobubbles were the reason. He then launched a company aiming to use the bubble formulation for agricultural applications, including improving plants’ resistance to disease. Revalesio later bought Wood’s company and further honed the bubble-creation technique. Currently its researchers generate RNS60, Revalesio's oxygen-filled nanobubble formulation, via a process involving something called Taylor-Couette-Poiseuille (TCP) flow. This is a mixing technique in which a saline solution is whisked rapidly under pressurized oxygen. Wood himself was diagnosed with amyotrophic lateral sclerosis in 2011. In 2012, following Revalesio’s completion of a Phase I safety study in healthy volunteers, Wood started receiving infusions of the bubbles under an FDA compassionate-use protocol. By the start of his treatment, Wood’s condition had already progressed to the point where he relied upon a ventilator, but he has since remained stable throughout nearly three years of treatment. Revalesio cannot conclude whether the plateauing of Wood’s disease is related to the nanobubble infusions.
Revalesio aims to treat people with inflammatory disorders, including multiple sclerosis and asthma, with RNS60. The plans are based on preclinical data suggesting that the bubbles calm microglial responses and boost regulatory T cells, which douse the overeager responses of other T cells that wreak havoc in autoimmune disease (see Khasnavis et al., 2012, and Mondal et al., 2012). Revalesio’s scientists later found that in addition to their effects on inflammation, the bubbles directly protected neurons. At SfN, Avik Roy, a researcher from Kalipada Pahan’s laboratory at Rush University in Chicago, presented a sampling of the data implicating the bubbles in neural regeneration.
Substance Behind the Bubbles
Roy and colleagues chipped away at the mechanism behind the phenomenon. They found that treatment with RNS60 boosted the growth, length, and density of dendritic spines on cultured hippocampal neurons. The treatment also upped the expression of NMDA and AMPA receptor subunits NR2A and GluR1, resulting in an increase in calcium intracellular signaling. In a gene-expression analysis, treatment with RNS60 turned on 62 of 84 genes associated with synaptic plasticity, including CREB; immediate early genes such as ZIF-268, Arc, and c-Fos; synapse-associated genes including PSD-95; and neurotrophic factors such as BDNF. Conversely, RNS60 quenched some genes known to dampen synaptic plasticity. PI3K inhibitors blocked all the gains in synaptic plasticity triggered by the bubbles, suggesting this signaling pathway was central to their broad actions.
The researchers then tried out RNS60 in 5xFAD mice, an aggressive AD model. Compared with those in normal mice, hippocampal neurons in 5xFAD mice had lower levels of key synaptic proteins, and reduced calcium signaling in response to stimulation with neurotransmitters, but two months of daily intraperitoneal injections of RNS60 reversed those effects. The researchers reported these data last July in PLoS One (Roy et al., 2014).
In the August 2 issue of the same journal, the researchers reported that RNS60 treatment rescued deficits in learning and spatial memory in 5xFAD mice (see Modi et al., 2014). RNS60 protected hippocampal neurons in these mice from apoptotic cell death, and shielded cultured neurons from death inflicted by Aβ. The bubbles also prevented hyperphosphorylation of tau in vivo in hippocampal neurons from 5xFAD mice, and downregulated the activity of GSK3b, a kinase implicated in phosphorylating tau, in cultured neurons. As with the synaptic plasticity boost, RNS60’s sway over neuronal death, tau phosphorylation, and GSK3b activity depended on the activation of PI3K, which was temporarily buoyed by the bubbles.
Revalesio’s recent findings mesh with those from Rodolfo Llinás’s lab at New York University in New York. An electrophysiologist whose bread and butter for 50 years has been the squid giant synapse, Llinás told Alzforum that he was at first intrigued by (and highly skeptical of) Revalesio’s findings. He decided to test the bubbles for himself. He reported that RNS60 improved synaptic transmission in squid neurons by mobilizing more vesicles to the synapse. Mitochondria in the treated neurons produced more ATP than usual. Llinás and colleagues reported that blocking ATP synthesis abolished all of RNS60’s positive effects (see Choi et al., 2014). Llinás’s lab has since found that RNS60 not only revs up ATP production, but enlarges mitochondria, according to a poster he presented at SfN. Rather than altering the kinetics of synaptic transmission, which could prove harmful, Llinás said that RNS60 simply injects more fuel into the energetically costly process. “I see it as a sort of ‘life optimizer’ rather than a drug,” Llinás said. “It makes everything run as efficiently as possible.” Llinás and colleagues also have a paper in press that reports similar findings in Xenopus giant oocytes.
How does it work? Llinás hypothesizes that the bubbles boost ATP production by delivering oxygen to the mitochondria. Normally, too much oxygen can be harmful, but Llinás said that he and his NYU colleagues Maxim Ivannikov and Mutsuyuki Sugimori have evidence that the bubbles cross the outer cell membrane as well as the mitochondrial membrane, then release the oxygen once inside the powerhouse organelle’s interior. In this way, the bubbles deliver their cargo without creating toxic free radicals in the cell, Llinás said. He thinks a majority of the effects both his lab and Revalesio’s researchers have reported stem from the extra ATP production triggered by this fuel injection.
Revalesio researchers are currently looking into the role of enhanced mitochondrial function in RNS60’s effects. However, for now they say their evidence points to the activation of PI3K as the major event that underlies most of the bubbles’ benefits. “Although it seems like there are various effects of RNS60, it actually has come down so far to a single signaling pathway: PI3K/Akt,” said Supurna Ghosh, Revalesio’s director of neurology research. “All of the effects we have seen so far in T cells, glial cells, or neurons can be abrogated by blocking that pathway.” How would the bubbles activate this pathway? “That’s the key question, and we really do not have an answer for it yet,” Ghosh said. PI3K can be activated from outside the cell via receptors on the cytoplasmic membrane, or from within the cell via other signaling molecules. “It’s possible that the bubbles enter the cell and activate it from inside, or possible that bubbles interfere with other proteins on the cell membrane, in turn activating PI3K,” she said.
Whether the bubbles’ benefits are derived from their physical interactions with proteins or from the way they deliver oxygen to mitochondria, it seems clear from the studies that both the bubble and the oxygen are key, Ghosh said. Control experiments with oxygenated fluids, or bubbles without oxygen, did not produce any of RNS60’s effects. This may dash the hopes of people hoping to glean some of the nanobubbles’ benefits by taking a few hits of oxygen at an oxygen bar (the establishments are growing in popularity and some claim to provide health benefits). Forcing oxygen into tissues by spending time in a hyperbaric chamber may also sound similar, however Watson said he thinks the bubbles’ structure is the key to their effects.
Regardless of exactly how the bubbles work, it seems likely that they are somehow delivered to the CNS, judging by experiments in mice, Ghosh said. Another recent study by the group reported that three hours after of being injected intraperitoneally, nanobubbles activated PI3K in the substantia nigra (see Khasnavis et al., 2014). Currently, there is no way to track the teeny bubbles in vivo, but Richard Watson, Revalesio’s chief scientific officer, said researchers are working on ways to do so by detecting gaseous oxygen. He estimates that the bubbles stick around in the body for about 18 hours before disintegrating.
RNS60 has so far proven safe in Phase 1 trials when injected intravenously or inhaled by healthy people or people with asthma (see clinical trials.gov; clinical trials,gov), and Phase 2 trials for both multiple sclerosis and asthma are about to begin, Watson said (see clinicaltrials.gov). Revalesio is currently devising inclusion criteria for its AD trial, which is expected to begin next year. It plans to enroll people with MCI or in the early stages of AD, and will use a combination of imaging and cognitive measures to monitor effects of RNS60.
At SfN, Watson was enthused by a presentation by Cynthia Lemere of Brigham and Women’s Hospital, Boston, which showcased GE180, a new PET imaging tracer for neuroinflammation (see Part 4 of this series). Watson hopes something like this could be a useful outcome measure in the RNS60 trial.
Lemere and Revalesio are hatching a plan to collaborate. Lemere plans to test RNS60 in AD mouse models, and use the GE180 tracer to measure its effects on neuroinflammation, she told Alzforum. Like most scientists, Lemere had her reservations about the bubbles at first. “It sounds far-fetched and bizarre, but the data looks really good and they’re doing rigorous science,” she said. While Lemere doesn’t believe that any one treatment will be a cure for AD, she said RNS60 could have potential, especially in combination with other therapies. “The fact that this has such broad application is really interesting,” she said. “I believe inflammation plays a big role in disease progression, so if you can quell the inflammation early in that process, you might be able to slow down neurodegeneration.”—Jessica Shugart




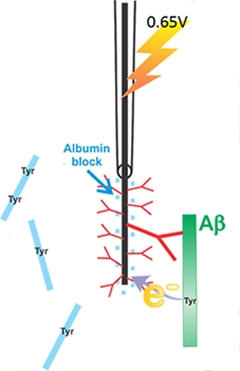

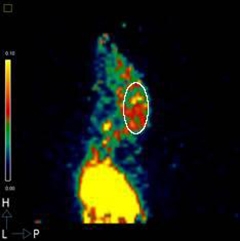

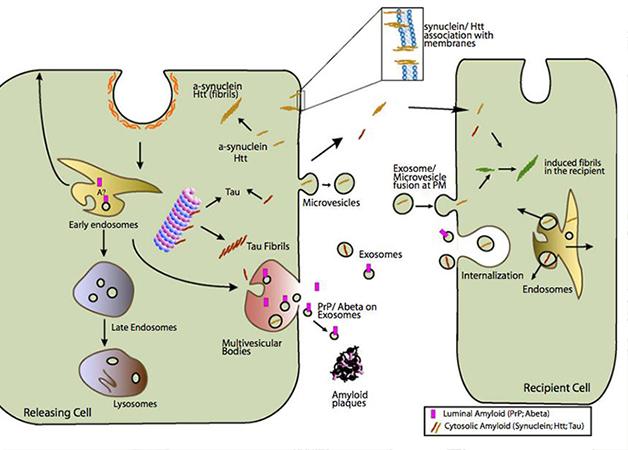
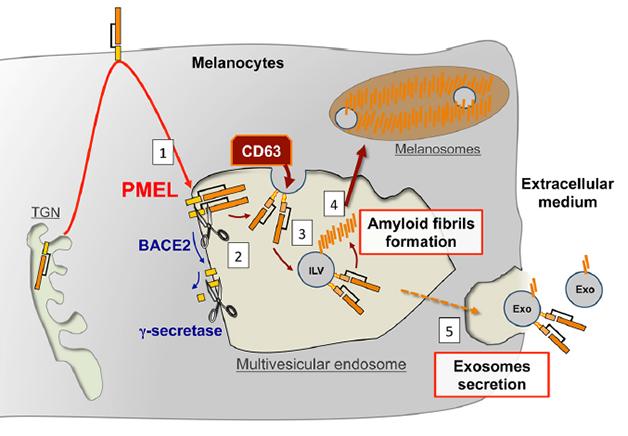
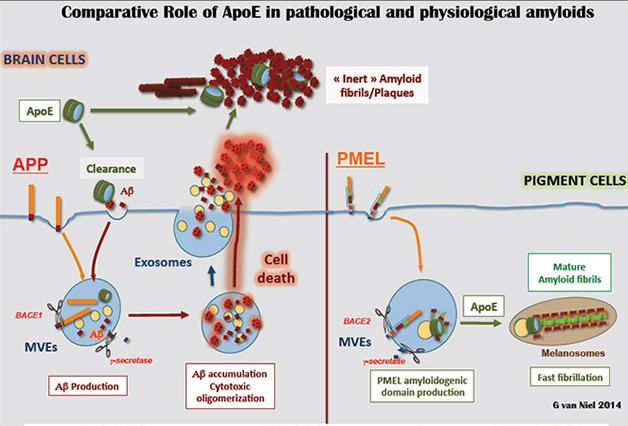



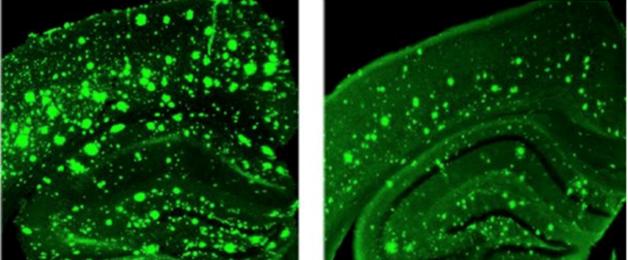
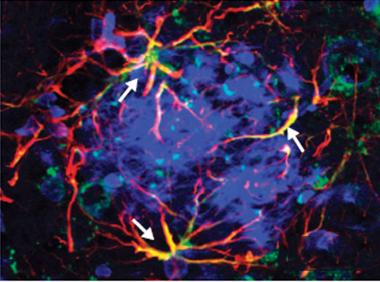

Comments
No Available Comments
Make a Comment
To make a comment you must login or register.