Tauists are having their heyday. First, stakeholders met last March in Las Vegas, Nevada, to discuss therapeutics for the tauopathy frontotemporal dementia. Days later, some 150 researchers flocked to the Gladstone Institute of Neurological Disease, San Francisco, to exchange their latest data at "Tau and Tauopathies: Pathogenic Mechanisms."
The Gladstone Institute teamed up with the German Center for Neurodegenerative Diseases, Bonn, to sponsor this inaugural meeting. Catch the hottest developments in this field with Esther Landhuis's unfolding coverage of the San Francisco conference and her now-complete coverage of the Las Vegas conference. Still hungry for more on tau? Download a copy of Lennart Mucke's in-depth review published in the May 12 issue of Neuron. [Image courtesy of Chris Goodfellow.]
San Francisco: Gladstone Institute Hosts Tau Powwow
Never mind that it featured 29 of the world’s top tau experts reporting the latest on their favorite protein. The recent symposium at the Gladstone Institute of Neurological Disease (GIND), San Francisco, ran less like a formal meeting and more like a two and a half-day journal club. Lured by sunshine, free bagels, and a focused agenda, 150 researchers packed Gladstone’s main auditorium 28-30 March 2011 for 20 hours of presentations and powwow on some of the field’s most fundamental questions. Further illustrating growing interest in tau, the conference occurred just prior to the appearance of an in-depth review on tau by conference co-organizer Lennart Mucke and colleagues in today’s issue of Neuron (Morris et al., 2011). In San Francisco, attendees discussed how tau goes awry, what else the protein does within neurons, and exchanged the newest cellular and animal data on various therapeutic approaches about how to translate a recent surge of scientific advances into new treatments for tauopathies such as Alzheimer’s disease and frontotemporal dementias.
“Tau and Tauopathies: Pathogenic Mechanisms” was sponsored jointly by GIND and the German Center for Neurodegenerative Diseases (DZNE). Founded in 2009, this government-funded consortium applies a translational approach to basic science research. Pierluigi Nicotera, who leads the DZNE, and Eckhard Mandelkow, who, along with his wife Eva-Maria, will leave the Max Planck Institute in Hamburg to join DZNE in Bonn, teamed up with Mucke to organize the workshop. It was the first of many the research centers intend to put on annually, alternating between San Francisco and Bonn.
The Gladstone Institute for Neurological Disease, San Francisco, hosted “Tau and Tauopathies: Pathogenic Mechanisms” in March. Image credit: Chris Goodfellow
The idea for the inaugural gathering arose last summer when Mucke, who directs the GIND, Nicotera, and Mandelkow were chatting about the tau field. “There had been pretty radical discoveries that challenged longstanding dogmas,” Mucke said. Among these provocative findings was the demonstration that removing tau protects neurons (see Roberson et al., 2007; Ittner et al., 2010; Yuan et al., 2008), that neurofibrillary tangles themselves might not be so dangerous (see de Calignon et al., 2010; Sydow et al., 2011), and that tau toxicity can be regulated post-translationally not only by phosphorylation, but also by other modifications such as acetylation (Cohen et al., 2011; Min et al., 2010).
“These are far-reaching findings, some controversial, that we thought would benefit from a face-to-face discussion,” Mucke told ARF. Interactive yet focused dialogue is hard to come by at big meetings, what with attendees dashing from one room to another trying to catch talks in parallel sessions. “We wanted to create a ‘time-out scenario’ where people could huddle and think about a topic carefully for days in a row,” Mucke said. The organizers encouraged speakers to reserve the last half of their 30-minute slot for Q&A, and most did. The topic apparently hit a nerve. Even though the organizers did not start planning in earnest until four months before the event, the speakers came and the event filled to capacity in a few weeks of open registration; more than a dozen companies sent representatives.
Bruce Miller of the University of California, San Francisco, opened with a clinical perspective on tauopathies. He reviewed their major symptoms and molecular pathologies (see ARF conference story), recapped recent work by UCSF colleague William Seeley showing that brain atrophy in tauopathies maps to functional networks (Seeley et al., 2009; Zhou et al., 2010), and presented sobering data suggesting a potentially large tauopathy epidemic in military veterans with traumatic brain injury (see McKee et al., 2009; ARF related news series on TBI; and ARF Live Discussion on long-term effects of concussions in athletes). In response to questions on biomarkers and trial design, Miller said that tauopathies proceed about twice as fast clinically and on brain imaging parameters than AD. A six-month period can reveal significant changes, he noted, a point that makes tauopathies attractive for clinical trials. Furthermore, a good tau imaging agent, like Pittsburgh Compound B (PIB) is for amyloid, would help researchers detect early, even pre-symptomatic, disease, Miller said. From Seeley’s work, “there is a sense that there may be a signal 20 to 30 years before people are symptomatic.” This is true for AD as well (see ARF related news story).
How Tau Turns Toxic
Several speakers addressed the issue of tau’s pathogenicity. Tara Spires-Jones of Massachusetts General Hospital, Charlestown, laid out three lines of evidence for why tangles may not be as toxic as commonly believed. First, using multiphoton imaging to peer into the brains of tauopathy mice, her lab has shown that tangles form in neurons after caspases get activated, not before (Spires-Jones et al., 2008; de Calignon et al., 2010). Tangle-bearing cells also stuck around a while after imaging, suggesting that the tangles themselves are not what kills the neurons.
Second, tangle-laden neurons appear functional in circuits. This came out of experiments using rTg4510 mice with doxycycline-regulatable expression of mutant human tau. These animals respond poorly to environmental enrichment, as judged by their blunted upregulation of the immediate early gene Arc in the cerebral cortex. However, when a six-week diet with doxycycline turned off the tau transgene, the mice’s Arc response returned to normal. Importantly, tangle-bearing and non-tangle-bearing neurons seemed equally responsive in this way.
The third piece of evidence calling into question the toxicity of tangles comes from array tomography studies examining the effect of these tau aggregates on mitochondrial transport in the same tauopathy mice. Initial experiments showed that the distribution of mitochondria was disrupted near tangles in the brains of 5.5-month-old rTg4510 mice. “Finally, something a tangle is bad for—it’s not letting mitochondria get where they need to go,” Spires-Jones recalled thinking at the time. However, once again, after the researchers suppressed transgene expression, mitochondrial distribution was restored even near tangles. “So it seems that the presence of aggregated tau is not what is causing the mitochondrial deficit,” Spires-Jones said.
Array tomography of rTg4510 mouse brain tissue stained for mitochondria (green), aggregated tau (Alz50, red), and tubulin (blue). Left panel shows a reduction of mitochondrial area fraction in Alz50-positive cells, which is reversed in animals with six weeks of doxycycline treatment to suppress tau transgene expression (right). Scale bars 10 microns. Image credit: Kathy Kopeikina
Lester (Skip) Binder of the Feinberg School of Medicine at Northwestern University, Chicago, Illinois, also hit on the relationship between conformation and toxicity. In collaboration with Scott Brady and Gerardo Morfini at the University of Illinois at Chicago, Nick Kanaan of the Binder lab studies how misshapen tau affects axonal transport in squid axoplasm—a convenient system for cell-free assays. “The squid has two big axons,” Binder said. “You can cut them out and roll out the axoplasm.” Even with membranes removed, transport maintains its polarity, allowing researchers to study motility and biochemistry right under the microscope. In this assay, tau monomers did not affect axonal transport; however, anterograde transport drastically slowed in the presence of tau aggregates, Binder reported. The researchers pinned this effect to a conserved stretch of 17 amino acids at the N-terminus of tau (Lapointe et al., 2008). Using an antibody they generated against synthetic tau oligomers, Binder and colleagues confirmed that this N-terminal region is exposed in oligomeric tau but not filamentous forms as exist in tangles. In the May 6 Journal of Biological Chemistry, Binder’s group formally published their characterization of tau oligomers shown last year at the Society for Neuroscience conference (see ARF conference story on Patterson et al., 2011). Binder’s and Spires-Jones’s observations bolster the emerging idea that neurofibrillary tangles may not be the most damaging form of tau, after all.
But if that is true, then how does monomeric tau morph into something that wreaks havoc on synapses and, eventually, learning and memory? This puzzle fuels ongoing research at the DZNE, where researchers led by Eckhard and Eva-Maria Mandelkow are unraveling connections between tau’s structure and its toxicity. Structural analyses are challenging because tau, like about 30 percent of all proteins, is intrinsically disordered. “One consequence of this is that the protein occupies a larger space in solution,” Eckhard Mandelkow said, noting that tau sweeps out a volume 27 times that of globular proteins with comparable weight. In solution, tau’s C- and N-terminal ends fold over the repeat domain, giving the protein a “paperclip” shape (Jeganathan et al., 2006; Jeganathan et al., 2008). Moreover, the scientists have shown that the regions of tau causing it to aggregate are small relative to the whole protein, and that the stretches associating with microtubules are the same as those needed for tau’s assembly into filaments. “The physiological and pathological functions of tau are encoded in the same parts of the molecule,” Eckhard Mandelkow said.
Nuclear magnetic resonance (NMR) studies (see Mukrasch et al., 2009) are yielding some insight into tau’s secondary structure. “Its conformations are never steady. All parts of the tau chain are mobile all the time,” Eckhard Mandelkow said. “You can think of tau as a collection of freely mobile amino acids held together by some bond.” Nevertheless, the researchers have figured out how to make aggregation-prone and non-aggregating forms of tau by tweaking residues that promote or discourage formation of β-sheets—a feature the Mandelkows believe is closely tied to tau’s toxicity.
As in the Aβ field, no one has put a finger on a particular toxic species of tau just yet. Even so, the studies lend hope that targeting tau aggregation could work as a therapeutic approach. Turning off mutant tau expression in the “pro-aggregant” mice allowed them to regain their synapses and cognitive function even while tangles remained. Furthermore, aggregation-blocking compounds can counteract toxic effects of aggregation-prone tau fragments in cell models (Khlistunova et al., 2006)—and potentially in a worm tauopathy model as well.
In San Francisco, Eckhard Mandelkow played a video clip of worms expressing human tau with the P301L and V337M mutations that cause frontotemporal lobar degeneration with parkinsonism linked to chromosome 17 (FTDP-17). These worms are unusually lethargic (Kraemer et al., 2003). In collaboration with Enrico Schmidt in Freiburg, Germany, the scientists also expressed their aggregation-prone tau fragment in these worms and found it worsened their tau pathology even further, to the point of paralyzing them, Eckhard Mandelkow said. Recent studies in his lab suggest that tau aggregation inhibitors can dampen this process.
Etienne Baulieu of INSERM in Paris, France, spoke about FKBP52. This is an enzyme—a peptidyl-prolyl isomerase that may change the secondary structure of tau, presumably affecting its neurotoxicity as well. Named for its ability to bind the immunosuppressant drug FK506, FKBP52 blocks microtubule formation through its binding to tubulin (Chambraud et al., 2007). When overexpressed in PC12 rat neuronal cells, the isomerase binds tau, keeps it from accumulating, and prevents neurite outgrowth induced by nerve growth factor (ARF related news story on Chambraud et al., 2010). When overexpressed in zebrafish, it shortens growth cone processes, and the posterior lateral line nerve develops abnormally. The enzyme also worsens motor development in P301L tau transgenic fish made by Dominik Paquet, Bettina Schmid and Christian Haass at the Ludwig-Maximilians University in Munich, Germany (see ARF conference story), Baulieu reported.
By confocal imaging, Baulieu and colleagues observed FKBP52 co-localizing with tau in brain cells embryonic wild-type mice and from postmortem brain samples of normal elderly, but not in samples from people with AD or various frontotemporal dementias. Though it is not clear how the FKBP52-tau association ties in with disease, the fact that these two proteins co-localize in normal brains, and not just in cell culture systems, hints at physiological relevance and jibes with FKBP52’s hypothesized protective action on tau, Baulieu said.
Meanwhile, researchers continue grappling with tau toxicity at the level of post-translational modifications and interacting proteins. For more on these developments, read Part 2.—Esther Landhuis.
Chambraud B, Sardin E, Giustiniani J, Dounane O, Schumacher M, Goedert M, Baulieu EE.
A role for FKBP52 in Tau protein function.
Proc Natl Acad Sci U S A. 2010 Feb 9;107(6):2658-63.
PubMed.
San Francisco: Making Tau Toxic—Post-translational Changes Galore
Given its correlation with Alzheimer’s disease progression, tau hyperphosphorylation has long commanded the field’s attention. Many labs have labored to identify disease-relevant phosphorylation sites on this microtubule-stabilizing protein, as well as the kinases and phosphatases responsible for those changes. Recent studies have shifted the focus, however. Some have uncovered acetylation, an altogether different post-translational modification, as potentially important for tau toxicity. Others are turning up potential targets for caspase-cleaved forms of pathological tau. These were among the topics scientists explored at a 28-30 March 2011 workshop, “Tau and Tauopathies: Pathogenic Mechanisms,” hosted by the Gladstone Institute of Neurological Disease in San Francisco and co-sponsored by the German Center for Neurodegenerative Diseases (DZNE) (see Part 1).
Acetylation—The New Force on the Block
Last year, researchers led by Li Gan at the Gladstone Institute reported that tau can be acetylated, potentially as a means of preventing its clearance by the ubiquitin-proteasomal system (ARF related news story on Min et al., 2010). Another study published earlier this year confirmed that tau is acetylated in cells, mice, and several human tauopathies. That report—by Virginia Lee and colleagues at the University of the Pennsylvania School of Medicine in Philadelphia—also confirmed that acetylation makes tau resistant to degradation and further suggested that acetylated tau is prone to fibrillization (ARF related news story on Cohen et al., 2011). First author Todd Cohen presented these data in San Francisco.
The data from these two labs suggest that reducing tau acetylation could be beneficial for treating tauopathies. In San Francisco, Gan said she wants to determine which acetylation sites are most relevant to AD; toward that end, efforts to generate monoclonal antibodies to specific epitopes are underway. The researchers are also making tauopathy mice with conditional deletion of SIRT1, an enzyme that can deacetylate tau. These tools should help address how acetylation affects tau function, Gan said. For their part, Cohen and colleagues have made an antibody that recognizes tau acetylated at K280, a site in the microtubule-binding region that is linked to frontotemporal dementia with parkinsonism-17 (FTDP-17). Deletion of this very lysine produces an aggregation-prone form of tau that causes synaptic and cognitive impairments when overexpressed in mice (Mocanu et al., 2008; Sydow et al., 2011). The UPenn researchers have confirmed that the antibody is sensitive and site-specific, and used it to examine acetylation of tau at K280 in several tau transgenic mouse models, and in postmortem brain tissue from AD patients (see below). Staining was low in hippocampal sections of PS19 mice carrying the P301S tau mutation, but much higher in PS19/PDAPP bigenic mice, Cohen reported.
Tau protein acetylated at K280 shows up in neurofibrillary tangles from brains of patients with Alzheimer's disease. Image credit: Todd Cohen
“These are interesting new developments,” noted Eckhard Mandelkow of the DZNE in an e-mail to ARF. “However, since tau is an open, natively unfolded protein, any change in cellular activity will likely have some post-translational impact on tau. As in the extensively documented case of phosphorylation, it will be difficult to prove that acetylation modifications are causative, rather than correlative.”
Perhaps the same can be said for other pathological forms of tau, where research is just beginning to establish links with cellular pathways. Gail Johnson of the University of Rochester, New York, spoke about her lab’s recent studies with a caspase-cleaved tau fragment (tau-D421) that accumulates in the brains of AD patients and is neurotoxic in a fly model for AD and related tauopathies (Khurana et al., 2010). Using mouse cortical cells with doxycycline-inducible expression of D421 truncated tau, or full-length tau as a control, the researchers determined that truncated tau was more resistant to autophagic degradation (Dolan and Johnson, 2010). Taking a page from other work suggesting mitochondria as a potential target for pathological tau (see review by Eckert et al., 2010), the Rochester team found that turning on tau-D421 expression in cortical neurons boded badly for their energy-producing organelles. Compared to cells expressing full-length tau, tau-D421-expressing cells had more fragmented mitochondria and membrane damage, as well as higher oxidative stress levels (Quintanilla et al., 2009). In experiments measuring oxygen consumption rates of cells in 24-well plates, tau-D421-expressing neurons had abnormally low basal mitochondrial respiration and respiratory reserve capacity, Johnson reported. She concluded with fresh-off-the-bench Westerns showing that caspase-cleaved tau, but not wild-type tau, interacts with mitofusin 2 (Mfn2). Among other functions, Mfn2 regulates mitochondrial transport and is a primary gene mutated in Charcot-Marie-Tooth-type 2A neuropathy. Johnson’s preliminary data suggest Mfn2 as a potential target for pathological tau.
Phosphorylation—So Familiar, So Mysterious
In addition to the new data on post-translationally modified and caspase-cleaved forms of tau, the conference included several talks on the historic tau alteration—phosphorylation—and its role in neurodegeneration. A pioneer in this research area, Khalid Iqbal, New York State Institute, recapped recent data solidifying the long-held claim that this modification is a key means for making tau troublesome. Previously, he and colleagues determined that protein phosphatase 2A (PP2A) accounts for about 70 percent of total tau phosphatase activity in the human brain (Liu et al., 2005), and that AD patients have decreased activity of PP2A and PP1 in their brains (Gong et al., 1993). Inhibitor-2 of PP2A, also called SET, moves from the neuronal nucleus to the cytoplasm, where it settles within neurofibrillary tangles in the brains of people with AD (Tanimukai et al., 2005).
More recently, Iqbal’s group expressed the C-terminal fragment of SET in the brains of rats using adeno-associated viruses. This caused abnormal tau phosphorylation, neurodegeneration, and cognitive deficits, solidifying the idea that phosphorylation of tau does, in fact, promote its neurotoxicity (Wang et al., 2010).
Continuing the phosphorylation theme, research led by Akihiko Takashima at the RIKEN Brain Science Institute in Wako City, Japan, focused on glycogen synthase kinase 3 (GSK-3), which phosphorylates tau, and evidence for its necessity in AD pathogenesis (see review by Takashima, 2006). In San Francisco, Takashima discussed a simple model in which AD is triggered by Aβ-related events and/or by aging factors that induce neurofibrillary tangles. He presented data suggesting that tau and GSK-3β deficiencies do not give rise to detectable symptoms early on, but cause problems as the animals age. In their analyses of GSK-3β and tau knockout mice, Takashima and colleagues found that younger animals did fine on spatial learning and memory tests, but showed deficiencies by 22-23 months of age. When the researchers tried to induce long-term depression (LTD), which normally activates GSK-3β, deficits showed up in both young and old GSK-3β and tau knockout mice. These data suggest that mice become more sensitive to LTD deficits as they age, Takashima said.
However, despite the long-standing focus on tau hyperphosphorylation, scientists do not yet have a handle on how this modification actually gives rise to disease (see review by Takashima, 2010).
Is phosphorylated tau functionally important? This question guides much of the work in the lab of Gloria Lee, University of Iowa, Iowa City, who presented some of her studies on tau as a signal transduction protein. Previously, Lee and coworkers had shown that tau interacts with the tyrosine kinases Src and Fyn in vitro (see review by Lee, 2005). Subsequent work, including analyses of tauopathy mice, suggests that some of these associations may be relevant to neurodegenerative disease (Bhaskar et al., 2005; Bhaskar et al., 2010).
Probing whether tyrosine-phosphorylated tau has a physiological role, Lee and colleagues found clues in a previously published microarray analysis of brain tissue from tau-deficient mice (Oyama et al., 2004). Cytoskeletal factors were notably absent from that paper’s top 10 list of mRNAs regulated by tau, suggesting that tau’s major function may not be cytoskeletal, Lee said. Instead, transcripts encoding the transcription factor FosB took the first two spots on that list. Lee’s team explored whether tau influences the signaling pathway leading to FosB expression by monitoring activation of AP-1—a protein that complexes with Fos to bind DNA—in tau-overexpressing and tau-deleted cell lines stimulated by nerve growth factor. The researchers found that AP-1 activity (judged by MAP kinase activation) was low in the tau-deficient cells, and that introducing wild-type tau into the cells invigorated the pathway. The effect held even when they put in a tau mutant that cannot bind microtubules, suggesting that “whatever tau is doing here, it doesn’t have to do with microtubules,” Lee said.
The scientists determined that a specific phosphorylation site on tau, Thr231, is critical for the protein’s ability to promote MAPK activation, and studies are underway to identify proteins that interact with tau further upstream in the AP-1/MAPK pathway, Lee said. In her working model, the road to neurodegeneration begins with abnormal phosphorylation of tau, which causes changes in cell signaling that lead to loss of cell cycle control. To see whether tau might, in fact, have a role in cell division, Lee’s lab is doing experiments in human prostate cancer cells, which, unlike mice, express all six tau isoforms that appear in the brain (see review by Souter and Lee, 2010).—Esther Landhuis.
Though amyloid-β and tau stand as the pathological hallmarks of Alzheimer’s disease, only recently have scientists gained insight into how the two could be linked mechanistically. Research from several labs suggests that tau acts downstream of Aβ. What’s more, recent studies have shown that animals lacking tau are impervious to the synaptic and cognitive problems normally brought on by Aβ overproduction, raising the notion of tau reduction as a potential therapeutic avenue. At the “Tau and Tauopathies: Pathogenic Mechanisms” workshop held 28-30 March 2011 at the Gladstone Institute for Neurological Disease in San Francisco (see Part 1), several speakers presented new data in this vein. Some of it complicates the picture, suggesting that tau reduction may not always be beneficial.
Early hints of tau’s essential role in Aβ’s dirty work came from cell culture studies showing that tau-depleted hippocampal neurons do not wither despite the presence of Aβ (Rapoport et al., 2002). Erik Roberson, while a postdoc in Lennart Mucke’s lab at the Gladstone Institute, confirmed those findings in vivo by showing that halving tau levels in APP-overexpressing J20 mice kept them from dying prematurely and helped them resist the Aβ-induced cognitive impairment (ARF related news story on Roberson et al., 2007). In addition, these scientists and others report that reducing tau protects APP mice against axonal transport defects (ARF related news story on Vossel et al., 2010) relieves their spontaneous epileptiform activity (ARF related news story on Roberson et al., 2011) and lessens Aβ-induced impairment of hippocampal long-term potentiation (Shipton et al., 2011). The tyrosine kinase Fyn may contribute to these effects, as a recent study finds that tau targets Fyn to post-synaptic N-methyl-D-aspartic acid (NMDA) receptors (ARF related news story on Ittner et al., 2010; see also review by Ittner and Götz, 2011).
At the meeting, Roberson, who now heads a lab at the University of Alabama at Birmingham, presented new data suggesting that the benefits of tau reduction may extend to APP mice that express a Fyn transgene but have low Aβ levels. Despite their low Aβ production, the APP/Fyn mice develop cognitive impairment, and almost half die within six months. However, putting these mice onto a tau knockout background rescued the mortality phenotype, Roberson said.
More evidence that tau reduction could be beneficial apart from Aβ came from Yadong Huang of the Gladstone Institute. When his group analyzed apolipoprotein E knock-in mice expressing either the human E3 or E4 isoform, the scientists found that E4 knock-in mice selectively lost GABAergic interneurons to an extent that correlated with the animals’ learning and memory deficits (Andrews-Zwilling et al., 2010). The GABAergic interneurons that did remain in the E4 mice had excessive tau phosphorylation, and the scientists were able to enhance their survival and prevent cognitive impairment by knocking down endogenous tau with microRNAs.
Tau also appears necessary for a pyroglutamate form of Aβ (pyroGluAβ) to inflict its harm on neurons. This is according to research by George Bloom, University of Virginia, Charlottesville, previously presented at the 2010 Society for Neuroscience meeting in San Diego, California. PyroGluAβ is a highly stable, neurotoxic species that arises when an enzyme joins the ends of a glutamate residue exposed by removal of Aβ42’s first two N-terminal amino acids. Bloom’s team, including former graduate student Justin Nussbaum and Probiodrug AG scientists led by Hans-Ulrich Demuth, showed that oligomers made by mixing Aβ42 with a touch of pyroGluAβ killed primary cortical neurons in culture more potently than either peptide by itself. However, these oligomers hardly posed a threat to cultured neurons from tau knockout mice, suggesting tau is needed for pyroGluAβ aggregates to become menacing (see ARF conference story). “Maybe amyloid is the trigger that gets pathology started,” Bloom said in San Francisco. “But once that trigger is pulled, tau must be present for Aβ to do its damage.”
Lest you think removing tau improves everything, data from Hana Dawson and coworkers at Duke University in Durham, North Carolina, gives reason for pause. When taken into culture, primary hippocampal neurons from tau-deficient mice mature with a delay (Dawson et al., 2001), and hippocampal neurons from tau-/- Map1b-/- mice do not extend their axons or migrate properly (Takei et al., 2000). Furthermore, crossing Tg2576 APP mice onto a tau knockout background gave rise to dystrophic neurites and axon degeneration—effects rescued by mating the APP strain to human tau transgenic mice. Traumatic brain injury (TBI) hastened the neuritic and axonal impairments, and the effects were intensified in APP mice on a tau knockout, relative to tau heterozygote background (Dawson et al., 2010) (see image below). This pertains to the emerging field of chronic traumatic encephalopathy, a tauopathy in concussed athletes and military veterans with TBI, Dawson noted in an e-mail to ARF (see also ARF Live Discussion and ARF related news story on TBI).
Among Tg2576 APP mice receiving traumatic brain injury at six months of age, those on a tau-deficient background (left) had more clumps of misshapen axons (aka “spheroids”) than tau did heterozygote controls (right). Image credit: Hana Dawson
Dawson also reported preliminary data showing that loss of tau worsens motor defects and brings on disease earlier in an ALS mouse model that overexpresses mutant SOD1 (A93G). To complicate matters, an earlier study found that tau deficiency did not matter much, good or bad, for the mouse central nervous system (Harada et al., 1994). “Removing tau can be a double-edged sword,” Dawson said. In discussion, scientists noted that the impact of tau reduction remains to be fully established. Some wondered whether strain differences may underlie the discrepancies between the various studies.
Dawson closed by noting that tau knockout mice probably have compensatory mechanisms. “How much worse would knockout of tau be in an adult?” she asked. This, as opposed to gene deficiency from birth, may more closely resemble a tau-reducing therapy started in people already afflicted with tauopathies.
Mucke hopes to address this issue using a new mouse model with regulatable neuronal expression of anti-tau microRNA that his group generated. Feeding the mice doxycycline keeps a lid on the microRNA, allowing endogenous tau to be expressed, but withdrawing doxycycline for just one week made an appreciable dent in tau expression. “So far, we see no obvious neurological deficits or other phenotypic changes,” Mucke reported. “Detailed analyses of these mice are in progress.” The data will be compared to those to come from the Roberson lab, which has generated conditional tau knockout mice using a Cre/Lox approach. In the meantime, Mucke and colleagues are screening RNA interference and drug libraries to identify tau-reducing reagents.
In a rare mention of “amyloid cascade hypothesis” at this tau meeting, Simon Lovestone of King’s College London, U.K., said “we have paid too much attention to ‘amyloid’ and not enough to ‘cascade.’” To address this shortcoming, Lovestone discussed how Dkk1—a Wnt signaling inhibitor that is increased in AD and in mouse models of neurodegenerative disease (Caricasole et al., 2004; Rosi et al., 2010)—may fit into this cascade. Lovestone and colleagues used whole-gene expression arrays to identify genes responsive to Dkk1, and to Aβ. The shared genes were induced in amyloidogenic transgenic mice (Tg2576) but not in a tauopathy model (hTau), Lovestone reported. This trend also held in their analyses of postmortem human brain tissue, supporting the idea that the shared gene set “sits neatly between amyloid and tau,” Lovestone said.—Esther Landhuis.
San Francisco: Tau—Time to Shine as Therapeutic Target?
At the “Tau and Tauopathies: Pathogenic Mechanisms” workshop held 28-30 March 2011 at the Gladstone Institute of Neurological Disease (GIND) in San Francisco (see Part 1), speakers voiced a sense of welcome relief, perhaps akin to foreigners enjoying the company of inhabitants of their native country in a faraway place. In this case, attendees did not all share a nation of origin, but, rather, bonded over a common research interest—the protein tau and its role in neurodegeneration. For years, the microtubule-associated protein has held the number two spot on the list of proteins implicated in Alzheimer’s disease, where amyloid-β has hogged the field’s focus. However, recent failures in AD clinical trials of amyloid-lowering approaches have left the field pondering. Researchers are asking whether they have gone after the wrong targets, relied too heavily on genetic models that may not capture sporadic disease, tested compounds in patients with confounding comorbidities, or whose disease is too advanced. Rising out of this contemplative frenzy is a growing sense that it’s high time to focus on tau—that this “runner-up” deserves more serious attention as a potential therapeutic target for AD and other tauopathies. This story will describe a slew of experimental treatment approaches, most at the cellular or rodent stage.
The theme of tau as an overdue target permeated a slide talk by Frank LaFerla of the University of California, Irvine. In a prior study, LaFerla’s group showed that setting off microglial inflammatory pathways worsened tau pathology in young triple-transgenic (3xTg-AD mice), and that blocking Cdk5 activation in the brain phagocytes could tone down these effects (ARF related news story on Kitazawa et al., 2005). The researchers found that tau pathology correlated with increased serum IL-1β in the transgenic mice, and wondered whether blocking signals through this inflammatory cytokine could possibly help. In experiments recently submitted for publication, the answer seems to be, yes. Unlike earlier approaches in the 3xTg-AD model that targeted tau but not did not improve Aβ pathology, treatment with anti-IL-1 receptor antibodies reduced both Aβ and tau arms of disease. It also lowered serum levels of key inflammatory cytokines and improved spatial memory in eight- to nine-month-old 3xTg-AD mice, LaFerla reported in San Francisco. To confirm and further explore the connection between tau and inflammation, the lab is crossing IL-1 knockout mice onto the triple-transgenic background.
LaFerla closed by recapping recent work his group did in collaboration with UC Irvine colleague Leslie Thompson, showing that four months of oral nicotinamide can boost cognition in 3xTg-AD mice (ARF related news story on Green et al., 2008). The treatment had no effect on amyloid-β levels or processing of its parent molecule, amyloid precursor protein (APP). As it turns out, the nicotinamide led to a dramatic reduction of Thr231-phospho-tau, as well as lower somatodendritic tau. The researchers have not yet looked for changes in axonal tau, LaFerla said.
Tau’s action in nerve projections is the focus of a different therapeutic approach under exploration by Kurt Brunden and coworkers at the University of Pennsylvania in Philadelphia. At the meeting, Brunden gave an update on his lab’s ongoing work with epothilone D—a brain-penetrant, microtubule-stabilizing compound that slowed axon degeneration and improved cognition when given to young PS19 tauopathy mice (Brunden et al., 2010; see also ARF conference story).
“It’s one thing to stop disease onset,” Brunden told the Gladstone audience. “But what about something more akin to treating patients who already have signs of disease?” Toward this end, Brunden reported preliminary data suggesting that epothilone had similar benefits for older PS19 mice in which tau pathology is well underway. Treated transgenics showed dose-dependent improvement in axonal dystrophy and microtubule density. They also had less hyperphosphorylated tau, as judged by AT8 immunohistochemistry using a system for Braak-like staging in mice (see Hurtado et al., 2010), and performed better in learning and memory tests. All told, the compound “given to PS19 mice either at or after onset of tau pathology improved a number of endpoints without safety or adverse events,” Brunden said. Bristol-Myers Squibb is planning a Phase 1 study of epothilone D, and would eventually like to test the compound in tauopathy patients, Brunden noted in an e-mail to ARF.
Grappling With the Basics
Other therapeutic approaches have taken shape as scientists ponder fundamental questions in the tau field. For instance, if tangles appear unnecessary for neuron death, and certain types of cellular dysfunction develop well before the tangles crop up (see Part 1), then what is it that makes tau neurotoxic? Some think post-translational modifications are key, while others place their bets on conformational changes. To Peter Davies of Albert Einstein College of Medicine, New York, an early leader in tau research, “both are probably true.” Davies presented recent data supporting each claim. In one study, he focused on c-Abl. The tyrosine kinase is fairly inactive in normal brain, but can phosphorylate tau, and associates with plaques and tangles in the brains of AD patients (Tremblay et al., 2010). In the March 2 Journal of Alzheimer’s Disease (Schlatterer et al., 2011), Davies and coworkers report on their new mouse with doxycycline-regulatable c-Abl activity in forebrain neurons. “If you express c-Abl in the forebrain, you get tau phosphorylation, cell death, and gliosis accompanying the cell loss,” Davies said. These data suggest that c-Abl may have a role in neurodegenerative disease, and could hold promise as a therapeutic target. Helping that case is the fact that compounds inhibiting this kinase (e.g., the cancer drug imatinib) are well tolerated in people.
Davies also reported preliminary results that speak to the second claim—that conformational changes are required for tau to turn toxic. Here, the researchers tested passive immunization for clearing tau pathology in JNPL3 transgenic mice expressing the P301L tau mutation that causes frontotemporal dementia. Starting at three months of age, the mice received four months of weekly intraperitoneal injections of a pan-tau monoclonal antibody, or a monoclonal (MC1) specific for a conformational tau epitope formed early in the aggregation process. In a pilot experiment, tau pathology dropped markedly in the MC1-treated group, as judged by total levels of insoluble tau in the brain, and pThr231 and pSer202 immunostaining in hippocampus.
“I’m stunned at how well this worked,” Davies said, though he noted that the mechanism remains unclear. Davies does not know what fraction of antibodies makes it into the brain, or whether an extracellular, prion-like form of tau may be involved. For now, the preliminary data suggest that the specificity of the antibody may be more important than affinity. At the 2010 Society for Neuroscience meeting in San Diego, California, Rakez Kayed of the University of Texas Medical Branch, Galveston, reported benefits from passive immunization of the same mouse (JNPL3) with a monoclonal antibody specific for tau oligomers. Kayed’s team gave eight-month-old transgenic mice a single hippocampal infusion of anti-tau oligomer antibodies, and found that the treatment cleared tau oligomers from CA1 and dentate gyrus neurons and improved the animals’ motor performance, compared to mock-injected controls (see ARF conference story).
Active and passive tau immunotherapy also helped tauopathy mice generated in the lab of Luc Buee at the University of Lille, France (Schindowski et al., 2006). Immunization with a phospho-tau peptide, or weekly injections of anti-phospho-tau antibodies, decreased tau pathology and prevented Y-maze memory deficits in this THY-Tau22 strain, which expresses mutant human tau (G272V and P301S) in the brain. It is notable in that these mice develop tau pathology as well as synaptic and cognitive impairments, but lack the motor symptoms that complicate behavioral testing of other tauopathy models. THY-Tau22 mice have clear learning deficits by nine to 10 months of age, some six months before neuronal loss is detected (Jeugd et al., 2011). Synaptic defects show up between nine and 12 months. The mice also have spontaneous seizures and gliosis that worsens with age, Buée reported. “In these mice, we can initiate long-term depression (LTD), like in normal mice, but cannot maintain it,” Buée said. LTD was restored in the THY-Tau22 mice by treating them with sodium selenate, a small compound that showed therapeutic potential in the pR5 and K3 tau mutant mouse models (Van Eersel et al., 2010).
Buée and colleagues also tested whether physical exercise could relieve tau pathology and cognitive woes in this tauopathy model. Starting at three months of age, the mice voluntarily ran about a kilometer each day for nine months. This regimen prevented behavioral defects in the Y-maze. “The running mice never develop this deficit,” Buée said. How was this possible? Buée and colleagues found no changes in transgene expression or in the gliosis that exists in this model in response to exercise; hence, disease persisted and exercise did not work through an anti-inflammatory pathway. Instead, the French scientists correlated the behavioral effect with decreased brain levels of phospho-tau at certain epitopes and with a preservation of cholinergic markers.
Interestingly, NPC1 and NPC2—cholesterol transport genes that are mutated in Niemann Pick’s disease—were upregulated after physical exercise, Buée said. Employing a viral gene therapy strategy that has reduced amyloid pathology in Alzheimer’s disease mouse models (Hudry et al., 2010), the Lille researchers injected into THY-Tau22 mice an adenovirus vector encoding neuronal cholesterol 24-hydroxylase. In preliminary studies, tau pathology was down and memory improved. Buée said he hopes to show more data in July at the International Conference on Alzheimer’s Disease in Paris, France.
Greg Cole of the University of California, Los Angeles, gave attendees yet another therapeutic strategy to chew on. This one involves feeding curcumin, a curry spice that Cole and colleagues previously showed can dissolve plaques and inhibit Aβ oligomerization in AD mice (ARF related news story on Yang et al., 2004). The researchers have now fed the spice to hTau mice for five months beginning at 14-15 months of age, when they already have tangle pathology, synaptic deficits, and cognitive impairment. Compared to untreated transgenic mice, hTau mice on the curcumin diet fared better in the Morris water maze, and expressed higher levels of excitatory synaptic markers NR2B and PSD-95, Cole reported. The treatment did not affect tangles, insoluble tau, or monomeric phospho-tau levels, but did suppress SDS-stable tau oligomers. The results are reminiscent of the cognitive recovery seen in tangle-bearing Tg4510 mice after tau transgene inhibition with doxycycline (ARF related news story on Santacruz et al., 2005), Cole wrote in an e-mail to ARF. The data “support tau oligomers as a target and curcumin as a pleiotropic drug capable of targeting pure tauopathy with late intervention,” he noted. Investigators at Jaslok Hospital and Research Centre in Mumbai, India, are recruiting AD patients for a Phase 2 trial of a highly absorbed, lipophilic curcumin formulation. Manufactured as a food supplement called LongVida by Verdure Sciences, Indianapolis, Indiana, this reportedly more bioavailable form of curcumin was originally developed by Cole and his wife, Sally Frautschy, also at UCLA.
Karen Duff of Columbia University, New York, spoke about a strategy that harnesses the macroautophagy system to tackle tauopathies. As shown on a poster by colleague Wai Haung Yu at the 2010 Society for Neuroscience meeting, eight-week treatment with the autophagy-promoting compound trehalose cleared tau aggregates and improved behavior in tauopathy mouse models with primarily motor (JNPL3 line) or cognitive (Tg4510 line) phenotypes (see ARF conference story). Preliminary functional magnetic resonance imaging (fMRI) done in collaboration with Scott Small, also at Columbia, suggests that trehalose can restore cerebral blood volume to normal in the Tg4510 mice, Duff said, noting that fMRI outcomes may be more applicable to human studies than rodent behavioral measures.
Another way of tapping intracellular degradation pathways to treat tauopathies came in a talk by Chad Dickey of the University of South Florida in Tampa. In addition to autophagy, cells use the ubiquitin-proteasome pathway to dispose of unwanted proteins, and heat shock proteins interact with molecules in both systems to help determine whether they will be dispatched in these ways. Dickey and others reported previously that inhibitors of the chaperone heat shock protein 90 (Hsp90) could promote degradation of mutated or hyperphosphorylated tau in mouse models of tauopathy (ARF related news story). In San Francisco, Dickey discussed his lab’s efforts manipulating another component of the chaperone system—Hsp70 proteins.
Collaborating with Jason Gestwicki at the University of Michigan, Ann Arbor, Dickey’s group screened for activators and inhibitors of Hsp70 ATPase activity and tested their hits in cell-based models and tau transgenic mice (Jinwal et al., 2009). Recently, the researchers have identified a new chemical scaffold, YM1, which is more potent and specific for the Hsp70 family than the earlier series of compounds. They have also found ways to tweak another variant, Hsp73 (aka Hsc70) to ready it for similar action. Normally, Hsp73 preserves tau (Jinwal et al., 2010). However, “we can make single mutations in this chaperone based on what we have learned using the Hsp70 ATPase inhibitors that force it to degrade tau instead,” Dickey explained in an e-mail to ARF. “With this information, we can home in on specific sites within Hsp73 that may be the most effective drug targets.” Targeting Hsp73 may be a good strategy because this variant is highly expressed in the brain and co-localizes with tau pathology, whereas other variants (e.g., Hsp72) do not.
Another potential therapeutic strategy centers around MSUT2, suggested Brian Kraemer of the University of Washington in Seattle. MSUT2 is the mammalian homolog of SUT2, a gene required for tau neurotoxicity in a transgenic worm model of tauopathy (ARF related news story on Kraemer et al., 2003). SUT2 overexpression caused the worms to rack up insoluble tau and lose neurons.
In a study published online in February, and appearing in print in this month’s issue of Human Molecular Genetics (Guthrie et al., 2011), the researchers found decreased levels of this protein in postmortem AD brain samples, but only in regions with tau pathology. Knockdown of MSUT2 in cultured tau-overexpressing human cells caused a dramatic drop in tau aggregation. All told, the data suggest that MSUT2 contributes to tau toxicity and aggregation, and that targeting this protein might be beneficial for tauopathies.
A big challenge for the field will be defining the tau forms that are most damaging, and isolating them from brains of patients. “We have to look in AD brain for these oligomeric species that a lot of people think are toxic,” Eva-Maria Mandelkow of the German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany, said during the open discussion at the end of the meeting. Tau researchers may be taking their cues from the Aβ field, where the discussion changed once researchers started isolating oligomeric species from human AD brain after years of studying various synthetic or cell-based forms (see ARF related news story on Jin et al., 2011; ARF related news story on Shankar et al., 2008).
Offering a different way of thinking, Jürgen Götz, University of Sydney, Australia, questioned whether toxic tau species even exist in the human disease brain, after all. “Maybe the problem is tau elevation, whereby tau ends up in the wrong place and interacts with proteins it doesn’t normally associate with, preventing them from executing their physiological functions,” he said. Götz discusses evidence for this concept, dubbed the “tau axis hypothesis,” in a recent review with colleague Lars Ittner (Ittner and Götz, 2011).
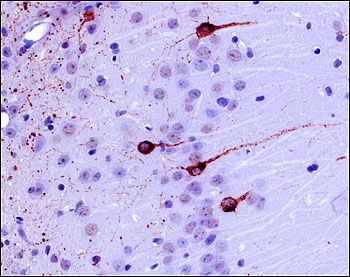
Ultimately, once researchers finger the forms of tau posing the greatest threat in the human brain, it will be important to determine whether these toxic species can seed and propagate disease, or whether this requires different forms of aggregated tau. These ideas arise from work led by Michel Goedert at the MRC Laboratory of Molecular Biology in Cambridge, U.K., in collaboration with Florence Clavaguera and Markus Tolnay at the University of Basel, Switzerland. Their studies showed that misfolded tau can travel from cell to cell in a prion-like manner in the brains of tau transgenic mice (see ARF related news story on Clavaguera et al., 2009; see review by Goedert et al., 2010). Goedert also reviewed recent data he reported last fall at the 7th International Conference on Frontotemporal Dementias in Indianapolis, Indiana (see ARF conference story). In those experiments, mice that received brain extracts from people with tauopathies went on to develop pathological deposits that resembled the human disorder (see image below).
Filamentous tau deposits (labeled with AT100 antibody) in the CA1 region of the hippocampus in an ALZ17 tau transgenic mouse 12 months after injection with human AD extract. Image credit: Florence Clavaguera
If misfolded proteins propagate in this fashion in human disease, “it suggests that tau pathology may start in just one cell, maybe even following the misfolding of a single tau molecule,” Goedert said. “Over a period of time, it would then spread from this one cell to other cells. The unpredictable nature of these early events means that it will be difficult to predict with great precision who will develop a tauopathy.”
Though many questions remain unanswered, attendees left energized about the surge of recent tau developments highlighted at the meeting. Some also voiced caution about relying too heavily on mouse models, noting how transgenic mouse studies have at times infused AD researchers with misguided optimism about preclinical approaches. Nevertheless, “we were really able to drill down deep,” Mucke said at the meeting’s conclusion. The next GIND/DZNE meeting will take place in Bonn, probably in May 2012, where the drill will penetrate the topic of synaptic pathophysiology in neurodegenerative disease.—Esther Landhuis.
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH.
Tau suppression in a neurodegenerative mouse model improves memory function.
Science. 2005 Jul 15;309(5733):476-81.
PubMed.
Jinwal UK, O'Leary JC, Borysov SI, Jones JR, Li Q, Koren J, Abisambra JF, Vestal GD, Lawson LY, Johnson AG, Blair LJ, Jin Y, Miyata Y, Gestwicki JE, Dickey CA.
Hsc70 rapidly engages tau after microtubule destabilization.
J Biol Chem. 2010 May 28;285(22):16798-805.
PubMed.
Jinwal UK, O'Leary JC, Borysov SI, Jones JR, Li Q, Koren J, Abisambra JF, Vestal GD, Lawson LY, Johnson AG, Blair LJ, Jin Y, Miyata Y, Gestwicki JE, Dickey CA.
Hsc70 rapidly engages tau after microtubule destabilization.
J Biol Chem. 2010 May 28;285(22):16798-805.
PubMed.





Comments
No Available Comments
Make a Comment
To make a comment you must login or register.