Organized around 10 major themes, this year’s annual meeting attracted almost 23,000 scientists. Sessions in the Neurodegenerative Disorders and Injury theme drew the largest crowd, featuring more than 2,700 of SfN’s total 14,000-plus presentations. Aβ, tau, and other protein pathologies dominated, but there was a renewed emphasis on gene therapy for neurological disorders. Tom Fagan brings you some highlights.
Not Just Blood Pressure—Dietary Salt Linked to Tau Phosphorylation
Too much salty food wreaks havoc on the cardiovascular system, raising blood pressure, damaging small blood vessels, and limiting perfusion into the brain. But is this why salt increases the chances of cognitive impairment? Not so fast. At this year’s Society for Neuroscience meeting, held October 19–23 in Chicago, Giuseppe Faraco from Costantino Iadecola’s lab at Feil Family Brain and Mind Research Institute of Weill Cornell Medicine, New York, reported that learning and memory deficits in mice chowing on a high-salt diet correlated with phosphorylation of tau, not with damage to the brain’s blood vessels. The study, published October 23 in Nature, links reduced nitric oxide in blood vessel walls to activation of kinases that modify tau. The findings present a new twist in the well-known link between cardiovascular disease and risk for cognitive decline.
Admittedly, at eight to16 times the norm, the amount of salt the mice consumed exceeds all but the very highest equivalents in which people might indulge. Still, researchers found the results thought-provoking. “However artificial the diet, this highlights that salt has effects independent of high blood pressure and that salt is a risk factor in its own right,” said Joanna Wardlaw, University of Edinburgh. Wardlaw thinks the mechanism may explain some clinical observations. “We’ve seen in studies of small stroke that despite treating high blood pressure, people continue to get worse clinically and on their brain scans,” she told Alzforum. “We need to think about the role of other common risk factors, including dietary salt.”
Li-Huei Tsai and Joel Blanchard, Massachusetts Institute of Technology, found the Weill Cornell group’s work fascinating. “They illustrate that neuronal cells and the cerebrovasculature have dynamic molecular and biochemical interactions that clearly influence neurodegenerative pathologies,” they wrote to Alzforum (full comment below). Faraco found the salt-induced reduction in nitric oxide (NO) boosted levels of p25, which activates the kinase Cdk5. Tsai has linked p25/Cdk5 to neurodegeneration (Dec 1999 news).
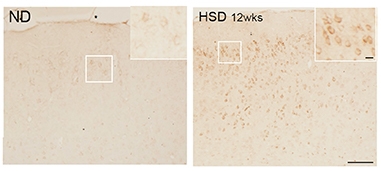
Pickled. AT8 immunostaining detects phosphorylated tau in the brains of mice fed a high-salt diet (right), but not in brains of mice on normal chow (left). [Courtesy of Giuseppe Faraco et al., Nature.]
The NO link most intrigued Zvonimir Katusic, Mayo Clinic, Rochester, Minnesota, as well. Susan Austin in Katusic’s lab found that knocking out endothelial nitric oxide synthase (eNOS) increases processing of Aβ precursor protein and impairs learning and memory, and most recently that it boosts p25 and phosphorylation of tau (Austin et al., 2010; Austin et al., 2013; Katusic and Austin et al., 2016). In Chicago, Austin reported that microglia from eNOS knockouts ramp up production of ADAM17, the primary sheddase for TREM2, and tone down production of the anti-inflammatory cytokine IL-10. “It appears release of NO by the endothelium is an important control mechanism for the brain,” said Katusic.
The plot gets thicker. The effect of high salt may not start in the endothelial cells of the brain, but in immune cells of the gut. Last year Faraco reported that a high-salt diet elicits a flood of interleukin-17 from T helper cells in the intestine. That IL-17 lead to a dearth of endothelial NO and impaired memory (Jan 2018 news). The IL-17 reduced cerebral blood flow by about 25 percent, but Faraco considers this insufficient to cause the memory impairment. Since tau pathology has been linked to cerebrovascular disease, he decided to see if a high-salt diet affected the microtubule binding protein.
Faraco put normal C56/Bl6 mice on a diet comprising 8 percent NaCl. This is 16 times the normal amount of salt in mouse chow; seawater is about 3.5 percent NaCl. The mice ate as much food as usual, but over the next 36 weeks, levels of phosphorylated tau rose. AT8 immunoreactivity peaked after 24 weeks, RZ3 immunoreactivity after 36 weeks. These antibodies recognize tau phosphorylation at serine 202/threonine 205 and threonine 231, respectively. Hyperphosphorylation of tau was detected in both male and female mice, and in mice on a 4 percent NaCl diet, albeit only AT8 staining in that case. Faraco found similar tau changes when he fed 8 percent salt to Tg2576 mice, which model amyloidosis. Levels of Aβ were unaffected.
What about neurofibrillary tangles? Faraco found none in any of the mice, but levels of insoluble tau released by formic acid did increase slightly in the cortices and hippocampi of mice on the high-salt diet.
In parallel with the tau phosphorylation, C57/Bl6 mice began having learning and memory problems. They struggled to recognize novel objects in their cages and had trouble finding the escape route in the Barnes maze. The deficits modestly correlated with AT8 binding in the cortex and hippocampus.
Was hyperphosphorylation of tau to blame? The authors tested this in two ways. They administered anti-tau antibodies to wild-type mice on high salt, and they fed high salt to tau knockouts. In both cases the animals performed as well as mice on normal chow, despite hypoperfusion of the brain, suggesting that indeed it was the tau that drove the cognitive decline due to the salt and not reduced blood flow.
Given Katusic’s prior data suggesting links between endothelial NO and tau phosphorylation, Faraco tested if he could stop the protein modification with L-arginine, a precursor in NO production. This suppressed both tau phosphorylation and the learning and memory deficits. In addition, elevated p-tau in eNOS knockouts could not be boosted further by high salt, supporting the idea that suppression of endothelial NO was behind the tau modification.
Delving more deeply into the mechanism, Faraco found that the salty food elevated calpain activity in the brain. Calpain cleaves p35 to p25; in keeping with this, the levels of the smaller peptide rose, as did activity of Cdk5, the tau kinase. All told, the data suggest that by triggering IL-17 production in the gut, high salt triggers loss of endothelial NO, which in turn leads to phosphorylation of tau and cognitive impairment.
Precisely how NO is suppressed remains to be seen. Katusic emphasized that the gas easily diffuses. Since cells in the brain are rarely more than 15 micrometers away from a blood vessel, NO could be an important signaling molecule. Faraco found no gross changes in astrocytes, microglia, or neurons in mice on high salt, as judged by GFAP, Iba1, and NeuN staining, but agreed it would be important to study downstream effects on these cells.
In her SfN talk, Austin reported that NO affected microglia more profoundly. In cultures of the cells from eNOS knockout mice, she found not only an increase in ADAM17, but also a decrease in cell surface TREM2. Mutations in this microglial receptor increase risk for Alzheimer’s and frontotemporal dementia. The sensor plays a central role in microglial homeostasis (Nov 2012 news; Oct 2012 news; Aug 2019 news). Austin also found that eNOS-/- microglia, either cultured or isolated from brain by cell sorting, make less TNFα and IL-10, pro- and anti-inflammatory cytokines, respectively, while at the same time ramping up phospholipase A2, which mobilizes arachidonic acid, a precursor for inflammatory molecules.
“We are slowly developing this concept that vascular mechanisms independent of perfusion affect cognitive impairment,” said Katusic. Tsai and Blanchard agreed. “Further unraveling these mechanisms will undoubtedly be a promising endeavor that will strengthen our understanding of how dietary habits influence susceptibility to age-related cognitive decline,” they wrote.
For his part, Faraco is using RNA-Seq to study what happens in the endothelial cells to reduce NO. “It will be interesting to examine interactions with other genetic and dietary risk factors, such as high-fructose or high-fat diets,” he said. He thinks it will be important to identify the tau species responsible for the effects on cognition. “We need to go much more deeply into the mechanism of neuronal dysfunction.”—Tom Fagan
Time to Try Again: Gene-Based Therapy for Neurodegeneration
Twenty years ago, researchers took fibroblasts from the skin of eight Alzheimer’s patients, engineered them to produce nerve-growth factor, and slid them into each volunteer’s basal forebrain. They hoped the neurotrophin would halt or slow the neurodegeneration that robbed them of their memories, indeed their lives. The gamble failed and since then, scientists have shown little zest for gene therapy in neurodegenerative disorders. That is changing. As evident at this year’s Society for Neuroscience conference, held October 19–23 in Chicago, gene therapy is back. Buoyed by success in treating spinal muscular atrophy in infants, scientists are flush with new ideas—and funding.
What was once considered risky, expensive, and unlikely to succeed is now seen by many as risky, expensive—and quite likely to succeed. A growing number of scientists think gene-based therapies may have the best chance of slowing, or even preventing, neurodegeneration, especially for disorders caused by mutations in a single gene. SfN hosted a press briefing on gene therapy, plus many projects are active throughout the field beyond those showcased at the conference. There was no breaking clinical trial news at the annual meeting, but the scope and challenges of such therapies were outlined at the briefing moderated by Rush University’s Jeff Kordower, Chicago, as well as a translational roundtable moderated by Asa Abeliovich, Columbia University, New York. Abeliovich recently co-founded Prevail Therapeutics, New York.
Going viral. Researchers are tweaking the capsid of adeno-associated viruses to optimize gene therapies for a multitude of disease. Shown here, AAV2.
From Zolgensma to Alzheimer’s?
If the failure of the nerve growth factor therapy tempered enthusiasm for gene therapy (Mar 2018 news), then the success of AVXS-101, aka Zolgensma, reignited it. Developed by scientists at Nationwide Children’s Hospital, Columbus, Ohio, and AveXis, Bannockburn, Illinois, AVXS-101 uses an adeno-associated virus to deliver billions of copies of the survival motor neuron 1 gene to the brain. A small pilot trial tested the therapy in babies with spinal muscular atrophy (SMA) Type 1, the severest form of this neurodevelopmental disease. Lacking functional SMN1, these infants face progressive muscle weakness. Most die before their second birthday; those who live need a ventilator to breathe.
In Phase 1, AVXS-101 dramatically improved motor function of 15 treated infants; all were living 20 months later when historical data predicted only one would survive. Twelve babies who received the highest dose grew stronger within months, most sitting independently and rolling over. They hit the highest score on a scale of motor function, whereas untreated babies deteriorated. By 20 months, two of the treated babies had begun to walk (Mendell et al., 2017). The Food and Drug Administration approved zolgensma in May 2019. At SfN in Chicago, Petra Kaufmann, AveXis, played videos of the first patients treated with AVXS-101. Some four years later, they are walking, running, and appear to be playing almost normally. A video of a little girl walking downstairs with nary a hint of having SMA Type I visibly moved the audience.
Scientists say it’s a game-changer. “It is really the tremendous success with SMA that has renewed interest in gene therapy,” said Clive Svendsen, Cedars-Sinai Regenerative Medicine Institute, Los Angeles. Speaking with Alzforum before SfN, Bart De Strooper, Dementia Research Institute, London, said the same. “The success in SMA patients of both gene therapy and antisense therapy has revived interest in the whole area,” De Strooper said. Nowadays, researchers tend to lump gene therapy and antisense therapy under one moniker, i.e., gene-based therapy. The SMA antisense therapy nusinersen also works in babies with SMA Type 1 and is FDA-approved (Nov 2016 news; May 2018 conference news). Unlike gene therapy, antisense therapy needs to be delivered indefinitely.
How About Neurodegenerative Disease?
At SfN, scientists outlined strategies for treating adults who face years of decline due to Alzheimer’s, amyotrophic lateral sclerosis, frontotemporal dementia, Huntington’s (HD) and Parkinson’s diseases (PD), or other synucleinopathies. Some are being tested in clinical trials, others are in preclinical development. Some target specific losses or gains of function, others aim to rescue dying neurons more broadly. Scientists also believe that working on rare childhood diseases of lysosomal storage may give them an opening to treat this common phenotype in age-related neurodegeneration, as well.
Just this October, an ApoE gene therapy trial started enrolling. Led by Ronald Crystal at Weill Cornell Medical College, New York, it will inject adeno-associated virus carrying the gene for ApoE2 into patients with early to late-stage AD who inherited two copies of ApoE4. The idea is to flood their brains with the protective allele of this apolipoprotein to try to counteract the effects of the risk allele. AAV-rh10-APOE2 will be injected directly into the subarachnoid cisternae of participants’ brains. The Phase 1 trial will recruit 15 patients with biomarker-confirmed AD. Beverly Davidson, Children’s Hospital of Philadelphia, has a similar ApoE2 gene therapy in preclinical development.
At SfN, Abeliovich detailed Prevail’s programs for forms of PD and for frontotemporal dementias that are caused by risk alleles. A trial has begun for a glucocerebrosidase-based gene therapy. The enzyme GCase is essential for lysosomes to function properly. People who have loss-of-function mutations in both copies of the GBA1 gene develop Gaucher’s, a lysosomal storage disease. The severest form starts in babies, most of whom die before age 2. Milder forms cause later-onset Gaucher’s, while heterozygous mutations in GBA1 increase risk for Parkinson’s, making restoration of GCase an obvious strategy for PD. Some researchers are trying to develop ways to boost activity of the mutated enzyme (e.g., Oct 2019 news), whereas Abeliovich and colleagues have constructed AAV-9 vectors to deliver normal GBA1 into the brain to restore GCase production.
In preclinical studies, the AAV9-GBA1 construct PR001 rescued both lysosomal and brain function in models of GCase deficiency and of Parkinson’s, Abeliovich said. In mice fed the GCase inhibitor conduritol β epoxide (CBE), PR001 injected into the brain ventricles beefed up GCase activity and reduced glycolipid accumulation, which is a sign that lysosomes are functional. A single dose worked for at least six months. Similar results were seen in a commonly used model of Gaucher’s that expresses the V394L GBA mutation and only weakly expresses prosaposin and saposins, lysosomal proteins that metabolize lipids. In these 4L/PS-NA mice, PR001 made increased levels of active GCase, fewer lipids accumulated, and the mice were more mobile on a balance beam. 4L/PS-NA mice also accumulate α-synuclein, the major component of Lewy bodies in PD and other synucleinopathies. In these mice, and also in A53T α-synuclein mice made worse with CBE, PR001 halved the amount of insoluble α-synuclein, Abeliovich reported at SfN.
In search of the right dose for humans, the scientists next turned to nonhuman primates. They injected PR001 into the cisterna magna in hopes AAV9 would broadly distribute throughout the brain. At the highest dose, 8 x 1010 capsids per gram of brain weight, exposure in the brain was similar to that seen in the mice. The virus permeated the spinal cord, frontal cortex, hippocampus, midbrain, and putamen.
Also in October, Prevail scientists began recruiting for a Phase 1/2 double-blind, sham-controlled trial to test this gene therapy in 16 people with moderate to severe PD, who have mutations in one or both copies of their GBA1 genes. Six patients each will receive a low or high dose of PR001A. Blood and CSF biomarkers to be analyzed at three and 12 months, and at follow-up, include GCase, lipids, α-synuclein, and neurofilament light chain. Participants will also undergo cognitive, executive, and motor-function tests and brain imaging. A Phase 1/2 trial of PR001 in neuronopathic Gaucher’s, which affects the brain and spinal cord, will start soon, Abeliovich said.
Other groups are boosting dopamine production in Parkinson’s by way of gene therapy. VY-AADC, developed by Voyager Therapeutics, Cambridge, Massachusetts, packages the gene for L-amino acid decarboxylase (AADC), which converts L-dopa into dopamine, in an AAV-2 vector that is delivered into the brain. Two Phase 1 open-label trials are testing safety and efficacy. Both the PD-1101 and PD-1102 trials use MRI to guide injections of the vector bilaterally into the putamina of 15 or 16 patients, respectively. According to preliminary results presented at the annual meeting of the American Academy of Neurology this past May, the virus penetrated half of the putamen and AADC activity, as judged by 18F-DOPA PET, increased by 85 percent in the latter study. Seven of eight treated patients reported improvement after a year, along with longer “on” time on L-DOPA, and shorter “off” time. Off time is the period when L-DOPA effects wear off and patients experience loss of motor control. RESTORE-1, a Phase 2 study of 42 patients, started in 2018 and will run to the end of 2020.
Long-Lived Gene Therapy. When a Parkinson’s disease patient died eight years after neurturin gene therapy, the trophin was still being expressed in their putamen (top left) and substantia nigra (bottom left), where it corresponded with tyrosine hydroxylase activity (right). [Courtesy of Jeff Kordower.]
Also in PD, Kordower and colleagues plan to re-evaluate neurturin-based gene therapy. Previously, the gene for this neurotrophin was delivered in an AAV2 vector into the brains of Parkinson patients in Phase 1 and 2 trials. This did not improve motor function. Even so, in Chicago Kordower showed that in two patients who died eight and 10 years later, the inserted gene was still expressing neurturin and that dopamine levels were higher on the injected than the contralateral side of the substantia nigra/putamen. “This shows us that long-term gene expression can be achieved in the human brain,” said Kordower (see image above). He believes that by focusing delivery with ultrasound, or tweaking the capsid itself, he may be able to generate enough gene expression to improve function.
Separately, AAV-GAD, a gene therapy for PD that showed promise in Phase 2 (Mar 2011 news) was acquired by MeiraGTx, New York, which will continue to develop it in the U.S. and Europe, according to founder Samuel Waksal (Nov 2018 news).
For its part, Prevail has a gene transfer construct for frontotemporal dementia in the pipeline, as well. Called PR006, it carries GRN, the gene encoding progranulin, on an AAV9 vector. GRN mutations cause familial FTD and, much like GBA mutations, do their dirty work via lysosomal dysfunction. In Chicago, Abeliovich reported that PR006 boosted progranulin release from neurons derived from FTD-GRN patients, nearly doubling their levels of mature Cathepsin D, the lysosomal protease that chops progranulin into granulins and indicates healthy lysosomes. In progranulin knockout mice, PR006 restored brain GRN expression and progranulin secretion into the CSF. Abeliovich said he expects a Phase 1/2 clinical trial in FTD patients to start in early 2020.
The biotech company Passage Bio, Philadelphia, is planning for clinical trials early next year with its AAV-GRN vector. MeiraGTx, New York, is banking on a different approach for FTD. They have developed an AAV carrying UPF1, which encodes regulator of nonsense transcripts 1. This protein helps clear out aberrant RNAs through a process call nonsense-mediated decay. MeiraGTx hopes this will restore homeostasis to RNA processing. AAV-UPF1 will be trialed for FTD and all forms of ALS bar those caused by mutations in SOD1. For SOD ALS, Novartis, Basel, Switzerland, and REGENXBIO, Rockville, Maryland, have a vector in preclinical testing.
For his part, Svendsen is taking a different approach. His lab tackles ALS with ex vivo gene therapy. The idea is to engineer clinical-grade human stem cells to produce glial-derived growth factor, and inject them into the spinal cord, much like the early NGF studies did in AD. Svendsen hopes the cells will churn out enough of the neurotrophin to protect spinal cord motor neurons. In a Phase 1/2a trial, 18 ALS patients have received these cells into one side of their spinal cords, such that each person serves as his or her own control. If this works, they would regain mobility only on the injected side. The trial finished in October; Svendsen expects results to come out in a few months. In a follow-up study, the scientists are trying to do the same with induced pluripotent stem cells. This would allow them to transplant autologous cells into patients, avoiding immune rejection
Other groups are deploying gene therapy as a way to improve immunotherapy, shield neurons from stress, or even generate neurons from astrocytes to make up for those lost to neurodegeneration.—Tom Fagan
Gene Therapies Enter Trials for Many Brain Pathologies—What about AD?
Buoyed by success in spinal muscular atrophy, scientists are once again devising gene-based therapies for neurodegeneration. Some are permanent, which makes proving their safety all the more challenging. Still, as shown at this year’s annual meeting of the Society for Neuroscience, held October 19–23 in Chicago, gene-based therapies for Alzheimer’s, Parkinson’s, frontotemporal dementia, amyotrophic lateral sclerosis, and some rare neurodevelopmental disorders are in clinical testing (see Nov 2019 conference news). Others are in preclinical development, as scientists are looking to suppress Aβ production, boost immunotherapy, or restore neurons lost to neurodegenerative disease.
Gong Chen and colleagues at Penn State University, in University Park, are pursuing the latter. “One reason so many clinical trials for AD have failed may be that too many neurons have already been lost,” Chen said. Previously, researchers in his lab reported that a single transcription factor, NeuroD1, can reprogram astrocytes into neurons, offering a potential way to replenish neurons in later stages of disease (Guo et al., 2014). This year, Chen reported that when spliced into an AAV9 vector and injected into the brain, NeuroD1 restored function after a stroke in both mice and nonhuman primates (Chen et al., 2019; Ge et al., 2019). NeuroD1 did that by creating not only new neurons, but astrocytes as well, as it prompts astrocytes to both divide and differentiate. The new astrocytes seemed to attract new blood vessels. “We are essentially regenerating new neural circuitry,” said Chen. Other groups are using similar, antisense oligonucleotide approaches to suppress transcription factors and steer progenitors toward becoming dopaminergic neurons to replenish those lost in in Parkinson’s disease (Jul 2019 conference news).
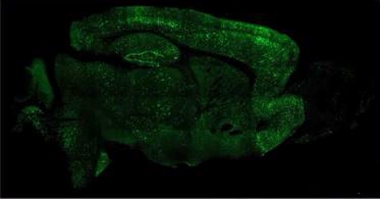
Could AAV-NeuroD1 work for AD? Chen and colleagues tried it in 5xFAD mice. “We regenerated millions of new neurons throughout the brain,” said Chen (see image below). The neurons survived at least eight months, while the number of reactive astrocytes fell. Mice treated with the vector learned and remembered better, finding a platform hidden in a water maze faster than untreated controls, Chen claimed. He is testing the vector in a nonhuman primate model of AD in China.
Cellular Conversion. Injected into the mouse brain, AAV-NeuroD1 generates millions of neurons (green) from astrocytes. [Courtesy of Gong Chen, Penn State University.]
Chen hopes the strategy will work in other diseases, as well. When injected into the spinal cords of mice carrying the G93A SOD1 mutation that causes ALS, AAV-NeuroD1 vectors restored motor neurons throughout the spinal cord, and improved motor function, he reported at SfN.
Others are trying to boost immunotherapies with gene-based tricks. Over the last 20 years, scientists have targeted a volley of therapeutic antibodies at various toxic proteins linked to neurodegeneration; alas, those immunoglobulins poorly penetrate the brain, usually at less than 1 percent of the injected dose. AAV offers a way of having them made right in the parenchyma, where they are needed.
One such therapy is an AAV9 carrying the genetic code for an anti-vascular endothelial growth factor antibody. Called RGX-314, it is in clinical trials for age-related macular degeneration. It was developed by REGENXBIO, Rockville, Maryland, which has partnered with Neuroimmune in Zurich to develop “vectorized antibodies” for neurodegenerative diseases.
In Chicago, REGENXBIO’s Olivier Danos said that this partnership will first target tau. His company holds patents on more than 100 novel AAV vectors, including AAV7, AAV8, AAV9, and AAVrh10, and is developing yet more variants based on tweaks to the capsid encasing the genetic information. Neuroimmune’s calling card is its strategy of screening healthy old people’s blood for naturally occurring antibodies that stave off diseases of aging. Aducanumab, a Phase 3 anti-Aβ antibody, was found that way (Oct 2019 news), as was the Phase 1 tau antibody BIIB076. Danos did not elaborate on which tau antibodies he will vectorized.
As for amyloid, Sergio Ferreira and colleagues at the Federal University of Rio de Janeiro developed Nusc1, a single-chain, variable fragment antibody whose DNA can be more easily packaged into AAV-9 than a full-size immunoglobulin. At SfN, Ferreira reported that Nusc1 binds to large Aβ oligomers in the human brain and neutralizes Aβ toxicity in cultured neurons, while having little affinity for Aβ monomers or fibrils (Sebollela et al., 2017). In vitro, hippocampal neurons infected with AAV-Nusc1 produced the antibody, which protected the cells from Aβ toxicity while control neurons bound Aβ oligomers and lost dendritic spines. When Maria Selles in Ferreira’s lab injected AAP/PS1 mice with AAV-Nusc1, it reversed memory deficits in novel-object-recognition tests. Ferreira hopes to test the therapy in clinical trials.
Other labs are starting to ramp up gene-therapy programs. “It feels really promising to be able to target the fundamental cause of a disease,” said Nick Fox, University College London. Fox believes that for some diseases, including familial AD, gene-based therapy could be the way of the future. “With single-gene disorders we have no doubt about the cause of the problem, and with these technologies we know we can go after it,” he said. Fox thinks a treatment for FAD will come before there is one for sporadic AD, which would be ironic, he said, given that people with FAD were told for years that they were not eligible for AD clinical trials.
Fox leads a gene-based research initiative at the UCL affiliate Queen Square Institute of Neurology. Funded by a £5 million donation from the Sigrid Rausing Trust, the Neurogenetics Therapies Programme plans to develop treatments for a variety of neurodegenerative diseases. “We have a strong desire to do the best for the patients and families we work with on a regular basis,” Fox said. The institute has been building cohorts of single-gene disorders such as Huntington’s, Alzheimer’s, and tauopathies since the 1980s.
“We want to create a genetic-therapy center where we can pool our expertise and resources, build a common infrastructure, share lessons learned, and formulate best practices,” said Fox. UCL’s Sarah Tabrizi and Cath Mummery have been collaborating with Ionis Pharmaceuticals, Carlsbad, California, to develop antisense oligonucleotides (ASOs) that target, and suppress expression of, mutant huntingtin and mutant tau, respectively. In Phase 1/2, IONIS-HTTRx halved the amount of mutant huntingtin in the CSF of carriers; licensed to Roche, this therapy is in Phase 3 (Mar 2018 conference news). The question with ASOs in general is whether they penetrate the parenchyma of the large human brain deeply enough to reach their targets. UCL is a site for Phase 1 testing of the tau ASO Ionis-MAPTRx.
Fox anticipates multiple preclinical projects at UCL, both AAV- and ASO-based, that build on the HD and tau programs to target AD, frontotemporal dementia, and other diseases. The UK DRI has slated £2 million for a gene-therapy program at King’s College London, where Chris Shaw will lead development of AAV-based vectors to treat ALS.
Other preclinical, gene-based therapies for AD include a project at Harvard University led by Connie Cepko to boost levels of Nrf2. This transcription factor protects neurons by turning on genes combating oxidative stress. At the University of California, San Diego, Mark Tuszynski, leads an AAV2 program to boost brain-derived neurotrophic factor production in the brain, and has completed a pilot trial in nonhuman primates (Xiong et al., 2015; Nagahara et al., 2018).
None of this is Easy
The researchers are well aware of challenges ahead. Some of the more-heterogeneous diseases, including late-onset AD, may be unsuitable. “We don’t have a good enough understanding of what would be a good target for sporadic AD,” said Bart De Strooper, U.K. Dementia Research Institute, London. Perhaps knocking down expression of APP or ApoE, but that might require suppression for five to six years, he speculated. The idea is to start with FAD and build from there.
How about regulating microglia? That might be further off, De Strooper believes. Microglia may be trickier to target via AAV than neurons, because the genetic material in AAV incorporates into an episome that stays put in the nucleus as long as the cell does not divide. That works well for postmitotic neurons; however, microglia are continually replaced, meaning the episome could be lost.
Repeat injections to replenish lost vectors are dangerous because the body mounts an immune response to the capsid the first time, hence a second injection could elicit a massive inflammatory response. “This is a major issue in the field right now,” said Petra Kaufmann, AveXis, Bannockburn, Illinois. Kaufmann said it might work to use a different vector the second time, or develop a way of first administration that allows a second, or to suppress the immune system. The latter is already being done. In trials of zolgensma, babies, who received up to 200 trillion virus particles per Kg, were also given prednisolone for 30 days.
It may be necessary to sidestep microglia to limit immune responses. “We want to minimize antigen presentation,” said Beverly Davidson, Children’s Hospital of Philadelphia. Even so, microglia might be targeted indirectly by “cross correction,” whereby a vector delivered to one cell type makes a protein that is secreted and taken up by another cell type.
Davidson has used this approach successfully to correct lipofuscinosis in dogs carrying loss-of-function mutations in the tripeptidyl peptidase 1 gene. This enzyme cleaves tripeptides from the N-terminal of proteins in the lysosome. In people, such mutations cause a form of Batten’s disease. When Davidson targeted ependymal cells with an AAV carrying the wild-type gene, the cells expressed the peptidase and released it into the CSF, whereupon it was taken up by parenchymal cells. Davidson showed videos of two 9-month-old Dachshunds with TPP1 deficiency. An untreated dog walked with extreme difficulty, tipping over every few steps, while the one treated with AAV-TPP1 ran around like a normal puppy (Katz et al., 2015). Davidson has also engineered AAV capsids to target endothelial cells, which then release the expressed protein into the lumen of the blood vessel. Of course, she noted, only proteins that are secreted by one cell type and taken up by another are suitable for cross correction.
Others warned that science needs to understand targets comprehensively before knocking them down with gene therapy. That is especially true for AAV. While antisense oligonucleotides will wash out once treatment stops, the AAV episome is permanent, at least in neurons. “This is a big risk,” agreed De Strooper. “I hope that at some point in the future we will have the means to control these vectors, maybe with promoters that are inducible and self-deactivating.” Clive Svendsen, Cedars-Sinai Regenerative Medicine Institute, Los Angeles, has used just such an approach in the engineered GDNF gene, which only turns on when doxycycline is present. “Being able to turn the gene on and off could be extremely important when this gene therapy is used in humans,” Svendsen said.
The challenges for neurodegenerative disease do not end there. Another one is getting enough of the vector into the brain and keeping it there. In Chicago, Danos showed that even when injected directly into the macaque central nervous system through the cisterna magna, AAV9 ended up in the thyroid, liver, spleen, and other organs within three days.
In macaques, AAV9 vectors caused some degeneration of dorsal root ganglia (DRG) accompanied by infiltration of immune cells, plus some axon damage in the spinal cord, though the animals’ behavior remained normal. In piglets, however, dorsal root ganglia lesions were accompanied by proprioceptive deficits and ataxia. On October 30, the FDA put a hold on STRONG, Novartis’ Phase 1 study of zolgensma in older infants, because of such animal data. And on November 12, the FDA halted, for a second time, a Phase 1/2 trial of SGT-001, a gene therapy for Duchenne muscular dystrophy developed by Solid Biosciences, Cambridge, Massachusetts. In a testament to the dangers of systemically administering AAV, this virus caused an immune response, loss of red blood cells, and kidney and lung damage in one of three patients who had received the therapy in October.
REGENXBIO, Davidson's, and many other groups are tweaking capsid proteins to improve targeting. For example, Sophie Mathiesen, University of Otago, Dunedin, New Zealand, described a modified capsid called PHP.eB. Unlike AAV9, it ignores the liver but infiltrates cells throughout the brain, at least in mice (see related Dec 2017 conference news). Still, Davison cautioned that, in some cases, pan-neurotropic viruses will not be good enough, and that they will have to be optimized to specific neuronal subtypes. Ideally, one wants to have ways to turn the viruses off, she said.
When asked about DRG side effects of his vectors, Abeliovich said that his team had specifically looked for this and found nothing. “We’ve tried multiple different doses in nonhuman primate models and looked specifically at DRG and white matter. At up to 1x1014 units/g of brain, we do not see any such effects,” he said. He thinks that the doses used in those other studies were so high that the DRGs became flooded with viral particles. “But these vectors are not all the same, and the most obvious difference is the cargo,” he said.
Danos agreed. “It could be the capsid, the expression level, toxicity linked to the CpG content of the genetic cargo … those are difficult to reproduce in different systems,” he said. “The reassuring message is that, as a rule, the vectors do not appear to be very toxic. But we need to look into it.”—Tom Fagan