This year, the AAIC conference showcased a field that is doggedly inching toward an effective anti-amyloid therapy while at the same time branching out from its usual ways of doing things. The antibody BAN2401 showed promising Phase 2 data, making it the latest in a handful of such therapeutics shown to robustly remove brain amyloid. (Hint: Crank the dose.) A fifth antibody, crenezumab, might remove CSF Aβ oligomers, a long-elusive species the field finally appears able to measure. Other scientists showed progress on capturing the decades-long disease process in deeper ways. They include measuring brain amyloid by way of a blood test, detecting neurodegeneration by way of blood neurofilament light protein levels, imaging synapses and—last but not least—new tests that catch subtle subtle cognitive deficits many years prior to the overt forgetting that used to be called symptom onset. (Hint: tau.) Non-amyloid-centered research is growing apace, supporting large initiatives to study the cardiovascular components of dementia, metabolic underpinnings of sex differences in AD, innovative drug-delivery approaches using ultrasound, and tech-based ways to support research and patient care.
The last full day of the Alzheimer's Association International Conference, which ended today in Chicago, saw scientists pack a room the size of an aircraft hangar in anticipation of a late addition to the scientific program. They came to see the data behind a tantalizing press release issued earlier this month, which had claimed that BAN2401, the anti-Aβ protofibril immunotherapy being developed by BioArctic, Eisai, and Biogen, reduced amyloid in early Alzheimer's disease and also slowed cognitive decline. The upshot? According to results presented in Chicago by Eisai's Lynn Kramer, the antibody appears to have done what it was designed to do. Over 18 months, fibrillar amyloid fell in all treatment groups compared with placebo; indeed, plaques melted by a whopping 93 percent in participants on the highest dose. This dose was reported to have reduced cognitive decline by 47 percent as measured by the ADAS-Cog, and by 30 percent on the ADCOMS, a new composite measure to detect early cognitive decline. At 856 participants with MCI due to AD or mild AD, this trial is the largest one yet to post both amyloid reduction and a downstream benefit on symptoms.
But wait. There's a catch. The data set is complicated. That's in part because this Phase 2B trial started out with adaptive randomization, which means that as participants accumulated, new enrollees were more apt to be put on doses that looked likelier to be effective. The trial featured a Bayesian statistical analysis at 12 months, and ended with a more conventional, aka “frequentist,” analysis at 18 months. That was complex, but the scientists had planned it that way.
Unplanned, however, was a curve ball regulators threw their way in July 2014, well over a year after the trial had started enrolling. Driven by safety worries about ARIA-E, the European Medicines Agency insisted that APOE4 carriers be excluded from the highest dose, 10 mg/kg infused biweekly. By that point in time, 74 carriers had been enrolled at that dose. What's more, the EMA demanded that all APOE4 carriers who had been on that dose for less than six months stop the trial, effectively kicking out 26 people who had no ARIA-E. This left 48 in the highest dose group. ARIA-E is a temporary inflammatory reaction typically seen within the first three months of Aβ immunotherapy; it is being studied intensely by most companies developing anti-Aβ antibodies. Most instances of ARIA-E occur within the first three months of dosing.
Kramer said that the trial sponsors argued against this restriction, to no avail. Paul Aisen from the University of Southern California, San Diego, said this regulatory request was odd, and difficult to understand. The EMA’s constraint likely compromised the adaptive randomization, said Andy Satlin, who initially led the BAN2401 program at Eisai and is now at Intra-Cellular Therapies in New York. This may have contributed to an outcome whereby the dose that proved to be the best received a lower number of participants than the second-best dose. In effect, the adaptive algorithm, which was increasingly leaning toward the highest dose as the trial went on, bumped up against the EMA’s APOE4 constraint, and a disproportionate number of participants ended up in the nearest dose. Suzanne Hendrix, a biostatistician and president of Pentara Corp., Salt Lake City, told Alzforum the restriction in effect biased comparisons with the placebo group. "The trial ended up with a different population in those two arms. That is not a true randomization anymore," Hendrix said.
Hendrix noted that among people with MCI and early AD, APOE4 carriers tend to decline faster on the ADCOMS, a cognitive composite she co-developed in hopes of picking up subtle changes at early disease stages (Wang et al., 2016). Outside this ADCOMS data, the idea of APOE4 speeding up progression is not established in the field, however. AD scientists do agree that APOE4 hastens amyloid deposition and brings on symptoms at a younger age, but they not agree on an effect on progression. “APOE4 carriers have an earlier onset, but their expected rate of decline is not different from patients with other APOE genotypes. However, there are conflicting reports in the literature on this topic,” wrote Lars Lannfelt of Uppsala University in Sweden, who made the mouse antibody that led to BAN2401 (see full comment below).
Despite the skewed randomization, statisticians and clinicians who gathered in the hallways after the presentation were cautiously upbeat. "Overall the results are positive and the amyloid effect is impressive," Hendrix said. "I believe this antibody works," Aisen agreed.
So do others. “In summary, there is dramatic amyloid lowering, with some apparent slowing in decline at the highest dose,” wrote Stephen Salloway of Butler Hospital, Providence, Rhode Island. David Holtzman of Washington University, St. Louis, considers the overall data “very promising” (full comment below), and Jeffrey Cummings of the Cleveland Clinic in Las Vegas thinks the results support target engagement and suggest a dosing strategy for phase 3. Randy Bateman of WashU wrote, “The field is clearly moving forward with the ability of a fourth drug to remove amyloid to a normal level, as measured by PET. Now with aducanumab, gantenerumab, and n3pg, BAN2401 has demonstrated reversal of amyloid plaques to normal levels, representing a milestone in the history of Alzheimer’s disease” (full comment below). Other commentators quibbled that the sponsors could have anticipated scrutiny over the APOE4 distribution and included that subgroup analysis in their AAIC presentation.
As the trial was enrolling, scientists conducted frequent interim analyses to steer new enrollees toward the doses that appeared early on to be most likely to work. This was done in hopes of making the trial more efficient (Satlin et al., 2016). It soon turned out that the action would be at the higher doses, and by the end of enrollment, 161 people were on 10 mg/kg biweekly, 253 people on 10 mg/monthly, and 247 people were on placebo. Only 52 people were in the 2.5 mg/kg biweekly dosing group, 51 in the 5 mg/kg monthly, and 91 people in the 5 mg/kg biweekly groups. Following the EMA request, the second-highest dose group ended up chock-full of APOE4 carriers, at 89 percent, compared with 30 percent in the highest dose group, and 70 percent in the placebo group.
At 12 months, a Bayesian analysis of the ADCOMS results estimated that the highest dose was 98 percent likely to slow decline. Separately, the trial's designers had previously set the criteria by which they would declare success at that time point to be an 80 percent likelihood of a clinically significant difference of 25 percent slowing of decline from baseline; the 12-month analysis calculated this likelihood to be 64 percent. The 80 percent was an awfully high bar, Hendrix told Alzforum, and may have been influenced by a push in the field at the time to reach for large effect sizes.
Bayesian trials had been debated in the Alzheimer’s field, but rarely put into practice. At AAIC, several commentators wondered if a simpler design could have worked as well. Hendrix expressed similar thoughts. "It's possible that with fewer dose arms, they might have met the primary endpoint at 12 months,” she said. On the other hand, Satlin said, the strong p values of the 10 mg/kg dose results may only have been achieved because the adaptive algorithm placed disproportionately more patients into this group than a conventional trial of the same size would have done.
So what were the results? Across the board, the antibody reduced amyloid in the brain. A time- and dose-dependent reduction saw PET SUVRs fall by up to 0.3 units in those on the highest dose, a 93 percent drop. Kramer said that on manual reads of the scans, 81 percent of treated patients went from amyloid-positive to amyloid-negative.
For the cognitive analysis, Kramer focused on the two highest doses. The 10 mg/kg biweekly group had a 47 percent reduction in cognitive decline as judged by the ADAS-Cog, and a 30 percent reduction on the ADCOMS. A 26 percent slowing on the CDR-SB was not significant. The 10 mg/kg monthly dose group—the one containing mostly APOE4 carriers—performed about midway in between placebo and the highest dose, showing a trend toward slower cognitive decline on the ADAS-Cog, ADCOMS, and CDR-SB but no statistical significance. The placebo group declined at a similar rate to placebo groups in recent large AD studies.
In a subgroup of patients who underwent spinal taps, CSF Aβ42 rose dose-dependently in all treatment arms, to more than 300-fold the level of the placebo group. Kramer said this was expected as the antibody pulled the peptide from plaques into the soluble fraction. Scientists generally believe that in the run-up to Alzheimer's dementia, CSF Aβ42 decreases as amyloid plaques deposit in the brain, sequestering it there. Kramer said studies were ongoing to determine how much of this Aβ was free versus bound to antibody. For CSF total tau, there appeared to be a slight reduction in the top two doses combined, though there was considerable scatter in the data.
And how about ARIA-E? As with other antibodies, it occurred in a dose- and APOE-dependent way, 48 times total across all groups, including two in placebo. Just shy of 10 percent of participants in the highest dose had an episode of ARIA-E, as did seven of that group’s 48 APOE carriers. Most ARIA-E was detected only on MRI, though five instances caused headache, visual disturbances, or confusion; two of those were in the highest dose, Kramer reported. In toto, this amounts to less ARIA-E than seen with gantenerumab or aducanumab.
Where does BAN2401 go next? "We view this as robust enough to approach regulatory authorities to discuss next steps in terms of additional trials or even breakthrough status," Kramer said in a press briefing before the main presentation.
In the general media, on social media, and among analysts, the results received the full range of responses, from an enthusiastic thumbs-up to a merciless drubbing over the uneven APOE4 carrier allocation. Both Biogen and Eisai’s stock prices dropped about 10 percent, but are starting to recover.—Tom Fagan and Gabrielle Strobel
An anti-amyloid antibody reduces levels of Aβ oligomers in the cerebrospinal fluid of most patients tested, according to late-breaking results presented at the Alzheimer’s Association International Conference in Chicago on July 25.
In collaboration with Genentech/Roche, Dennis Selkoe and Dominic Walsh, both at Brigham and Women’s Hospital, Boston, used an oligomer-specific ELISA assay they had developed to quantitate soluble Aβ assemblies in CSF from participants in two Phase 2 trials of crenezumab. After 69 weeks of treatment with this immunotherapy, oligomer levels had fallen almost in half in the majority of treated patients, Selkoe reported.
“The results strongly suggest principal target engagement of crenezumab with oligomeric Aβ,” said co-author Tobias Bittner of Roche. The results mark the first time that researchers have measured Aβ oligomers in clinical samples from trials of antibody therapies. Finding a decrease with treatment is encouraging, said Selkoe. “Theoretically, that could lead to therapeutic efficacy,” he told the audience. The result also suggests that crenezumab is well-positioned to test the Aβ oligomer hypothesis, Selkoe said.
That hypothesis holds that the neurotoxicity of amyloid comes mainly from small, soluble oligomeric assembles of Aβ (oAβ), which exist in equilibrium with larger amounts of insoluble Aβ in plaques. Despite growing interest from pharma in anti-oligomer therapies (Dec 2017 conference news), the toxic species have proved difficult to isolate and characterize, never mind to detect and quantitate in vivo.
Selkoe and Walsh recently demonstrated that oAβ extracted from postmortem brain tissue of AD patients represents a minority of the Aβ species in brain, but accounts for the majority of the toxic activity toward neurons (Jul 2018 news). They previously developed a high-sensitivity ELISA using their 1C11 antibody, which shows 30,000-fold selectivity for oAβ over monomeric Aβ (Yang et al., 2015). The optimized assay detects as little as 0.15 pg/ml of the oligomers, giving within-sample variances of less than 20 percent, and most often less than 15 percent, Selkoe told Alzforum.
The CSF for the new study came from the ABBY and BLAZE Phase 2 trials, both of which tested crenezumab in people with mild to moderate AD. In all, Selkoe’s lab tested CSF from 104 patients, drawn at baseline and again at 69 weeks into the trial. Concentrations of oAβ were quite low, with most samples reading out less than 10 pg/ml. Six of the samples tested below the 0.15 pg/ml limit of detection at baseline and were excluded. The investigators analyzed the remaining 98 baseline/endpoint pairs, which included 28 samples from the placebo group, and 35 each from people who had received the antibody by intravenous injection of 15 mg/kg every four weeks (high dose) or subcutaneous injection of 300 mg every two weeks (low dose). The testers were blind to patient treatment status.
According to the results presented at AAIC, there was no difference in oAβ between the placebo and treatment groups, or between the baseline and endpoint. Nonetheless, most of the people in the two dose arms posted a decrease in CSF oAβ over the course of the trial. In total, 86 percent of the intravenous group and 89 percent of the subcutaneous group registered a decline. In the placebo group, half of the subjects increased their oAβ and half decreased. “We were struck when we broke the blinding, that most of the patients who had been on crenezumab had a substantial decline in oligomer levels,” Selkoe said.
Treatment with crenezumab caused a reduction in the median oligomer concentration by 43 and 48 percent from baseline to week 69 in the two respective dose groups. As a result, 20 and 14 percent of participants in each arm fell below the 0.15 pg/ml detection limit of the assay. That exceeded the change in the placebo group, whose median value slipped, too, but just 13 percent, with no patients dipping below the assay detection level.
Why would the placebo group change? Selkoe noted that oAβ levels vary from person to person, and attributed their change to biological variation in a low-abundance analyte. “The intra-patient variance, i.e., same person on different days or months, and inter-patient variance in oAβ levels is considerable. I don’t think that a mean 13 percent change in the concentration of oAβ in either direction is surprising, as there may be quite a lot of dynamics of the low amounts of soluble oligomers in human CSF,” he wrote to Alzforum. “The key message is that despite biological variation, about 90 percent of the time crenezumab treatment lowers oAβ levels over 18 months,” Selkoe said.
In contrast, treatment increased soluble, monomeric CSF Aβ. Values before treatment ranged from several hundred to 1,000 pg/ml, and in three-quarters of the treated group, soluble Aβ rose, with a median change of 9–10 percent. That treatment response jibes with previously presented trial data (Cummings et al., 2018; Dec 2014 conference news). The Phase 2 BAN2401 trial that was also presented at AAIC posted an increase of more than 300 pg/ml CSF Aβ42 in the highest-dose group, but gave no percentages (Jul 2018 conference news). This antibody binds Aβ protofibrils, a larger species than Selkoe and Walsh focus on, but smaller than fibrils.
Measuring minute quantities of oligomeric Aβ is a challenge. “The assay is certainly pushing the limit of what can be measured with an immunoassay and is therefore less precise compared to the Elecsys Aβ42 assay,” Bittner wrote to Alzforum. “It gave reproducible results if the baseline and week 69 samples were measured on the same plate,” Bittner added.
Generally speaking, the target of a therapeutic antibody can be difficult to quantify by immunoassay in the presence of that antibody, because the therapeutic and the assay antibody could compete for the same target. In this case, the reduction in oAβ did not seem to be due to crenezumab in the CSF of treated people interfering with the assay, Selkoe believes. As a control, the investigators spiked crenezumab into human CSF, and this did not affect the ELISA’s ability to detect oAβ in subsequent testing.
This late-afternoon session on the fourth day of AAIC was sparsely attended, but some scientists liked the work even from afar. David Brody at Uniformed Services University of the Health Sciences, Bethesda, Maryland, works on purifying and measuring oligomeric Aβ (May 2016 conference news). Brody wasn’t at AAIC, but reading the abstract inspired him to rattle off research ideas. “The data in the abstract is exciting and nicely presented. This may be just the kind of target-engagement biomarker the field needs. There are, of course, many questions: How do CSF levels of soluble Aβ aggregates relate to those present in the brain and, most importantly, to those engaging their targets? Do changes in CSF levels of soluble Aβ aggregates correlate with changes in cognitive function? Is the approximately 50 percent reduction in CSF soluble Aβ aggregate levels the maximum possible response, or would higher doses of crenezumab produce even greater reductions? Do other therapeutics that have been unsuccessful in Phase 3 also lower CSF levels of soluble Αβ aggregates?”
Susan Catalano of Cognition Therapeutics, Pittsburgh, was there, and raised many of the same questions (see full comment below). “The results presented at AAIC are exciting and have major implications for the AD therapeutics field,” she wrote in an email. However, she cautioned, “The amount and even the direction of change in oligomer concentrations in the CSF that would reflect effective oligomer target engagement in the brain is still emerging.” Going forward, it will be important to correlate such measures with synaptic structure and function, as well as with cognition, she wrote.
What next? Bittner noted that Roche intends to replicate Walsh and Selkoe’s testing in the ongoing, fully enrolled crenezumab Phase 3 CREAD studies that test a fourfold higher dose of crenezumab in early AD patients.
How about the ongoing Alzheimer’s Prevention Initiative trial of crenezumab in Colombian presenilin1 E280A mutant autosomal-dominant AD families? Currently there is no plan to test their CSF samples, although Bittner and Selkoe agreed that would be worthwhile. “I think oligomeric Aβ should be looked at in all amyloid-targeting AD trials that have a significant number of CSF samples. Roche and we are working to try to make the assay widely available, though this will take some time,” Selkoe said.—Pat McCaffrey
PET Ligand Lights Up AAIC, May Detect Synapse Loss in AD
Among Alzheimer’s disease pathologies, synaptic loss correlates best with symptoms, but until now researchers have had no way to measure it in living people. That may be changing, with a new PET ligand that binds synaptic vesicle glycoprotein 2A (SV2A). At the Alzheimer’s Imaging Consortium symposium on the day before this year’s Alzheimer’s Association International Conference, July 22–26 in Chicago, Richard Carson of Yale University, New Haven, Connecticut, reported that the probe, UCB-J, bound significantly less in the hippocampi of people with Alzheimer’s disease than in controls. Poor retention of the ligand correlated with flagging memory, he showed. The work was published in the July 16 JAMA Neurology, and Alzforum previously reported some of the data when Christopher van Dyck, also from Yale, presented last fall at the Clinical Trials on Alzheimer Disease meeting in Boston (Dec 2017 conference news).
At AAIC, multiple attendees told Alzforum the work was a highlight of the imaging consortium session. “PET measurements of presynaptic and postsynaptic density would greatly advance the scientific study of Alzheimer’s disease and related disorders," said Eric Reiman, Banner Alzheimer’s Institute in Phoenix. "The Yale group has developed this promising 11C-labeled ligand to assess presynaptic density. They have begun to develop an 18F-labeled ligand that would advance its use by other centers, and they have begun to test these radioligands in rigorous and thoughtful ways," he wrote in an email to Alzforum (see complete comment below).
Rik Ossenkoppele, Vrije University, Amsterdam, called the work impressive and important. “The availability of a PET tracer that allows detection of structural synaptic alterations has important potential implications for clinical studies,” he wrote (see comment below).
Others were more reserved, asking if the probe truly bound synapses or some other vesicle fraction in the brain. Carson said that has yet to be worked out. “We need to understand the relationship between vesicles and synapses, and how it is affected by vesicle metabolism, including exocytosis and recycling, even synaptic activity,” he said. One audience member questioned if the distribution of synapses in the human brain maps to UCB-J binding. Carson replied that electron microscopy and other work to address this is ongoing. Some were concerned that the binding pattern varies from that seen with FDG, a PET ligand that measures glucose metabolism and is widely considered a surrogate for synaptic activity.
Still, the overall mood at AAIC was that this ligand could provide useful information about the brain. Developed by Carson, 11C-UCB-J binds with high affinity and specificity to SV2A, a universal marker of presynaptic vesicles. SV2A distribution closely matches that of synaptophysin, which is commonly used as a marker for synapses in postmortem human tissue. Previously, Carson and colleagues reported that it detected synaptic loss around focal seizures in people with epilepsy (Jul 2016 news).
In the AD study, Ming-Kai Chen, who works in Carson’s group and is first author on the JAMA Neurology paper, measured brain binding of 11C-UCB-J in 10 people who had mild cognitive impairment or dementia and who had tested positive on amyloid PET scans. He compared them to 11 cognitively normal, amyloid-negative, age-matched counterparts. The investigators focused their attention on the hippocampus, where they expected early synapse loss due to the degeneration of projections from the entorhinal cortex. They calculated relative binding of UCB-J using centrum semiovale white matter as a reference region, because it poorly binds the ligand.
The Alzheimer’s patients bound 41 percent less tracer in the hippocampus than did controls. That difference held even after correction for hippocampal atrophy. Chen also found lower binding in the entorhinal cortex, but that was explained by loss of tissue volume.
Postmortem examination of AD brains reveals widespread synaptic loss in the cortex, yet in this study, PET detected no difference between cortical binding of 11C-UCB-J in patient and control cortices. Why? The authors speculate the study was too small, or that most of the AD patients were at an early disease stage and had not yet lost cortical synapses. Another possibility is that remaining synapses had grown to compensate for those that had withered. This phenomenon, called synaptic hypertrophism, has been documented in mild AD (DeKosky and Scheff, 1990). Ossenkoppele wondered if the patient-selection criteria, which required poor scores on an episodic memory test, had enriched for people with primarily limbic pathology, i.e., hippocampal atrophy. Chen told Alzforum they plan follow-up studies, including postmortem analyses, with more patients in later stages of disease.
In an editorial accompanying the paper, Elizabeth Mormino of Stanford University, Palo Alto, California, and William Jagust, of University of California, Berkeley, raised the FDG issue. They point out that the decrease in UCB-J binding observed only in the hippocampi of people with AD differs dramatically from the more widespread cortical hypometabolism detected using FDG-PET. “This is surprising, because although hippocampal synaptic loss is severe, other brain regions, such as the frontal cortex, entorhinal cortex, temporal cortex, and cingulate cortex, have been reported to show a similarly high synaptic loss in postmortem studies that may even predominate in the presynaptic terminals measured with an [sic] synaptic vesicle glycoprotein 2A ligand,” they write.
Others have puzzled over this FDG/UCB-J dichotomy, as well. “Since FDG PET has been suggested to provide information about the density, activity, and metabolism of terminal neuronal fields and/or peri-synaptic glial cells, it will be important to understand the biological basis for these differences,” noted Reiman. Victor Villemagne, University of Melbourne, Australia, wondered why UCB-J did not detect synapse loss in cortical areas. “Is function (FDG) more sensitive than synaptic density?’” he asked (see comment below).
Chen and colleagues did detect less blood flow in some cortical regions of the MCI/dementia patients compared with controls. These were the regions where FDG-PET wanes in people with AD, but this blood flow reduction seemed to have no effect on UCB-J binding. Chen said this underscores that the two probes do not measure the same thing—where the FDG-PET signal provides a composite of glucose uptake by pre- and postsynaptic neurons and nearby glia, UCB-J provides a snapshot of synaptic structure and integrity. For this reason, he said, “We do not think that UCB-J-PET will replace FDG-PET, but instead that the two will complement each other.”
In a poster at AAIC, co-author Adam Mecca presented results from a preliminary analysis where he directly compared UCB-J and FDG-PET in nine people with MCI or AD, and 11 controls. He reported the expected pattern of FGD-PET hypometabolism in multiple cortical regions in the patients, and reduction of UCB-J binding in the hippocampus, but again no other regions. The investigators found a correlation between FDG-PET and UCB-J-PET signals in the hippocampus across both patients and healthy controls.
Other labs plan to test the probe. Bradley Christian, University of Wisconsin, Madison, said his group synthesized the tracer in-house, and completed animal testing before applying for FDA approval for human studies.
Carson and colleagues are developing 18F probes, which would expand use beyond centers that can make their own supply of the short-lived 11C version. Here things get tricky. UCB-J contains three fluorines. “We have labelled it with F18, but the chemistry is terribly difficult,” Carson admitted. Instead, the researchers have turned to making derivatives that contain a single fluorine. Two, called SDM8 and SDM2, look promising, said Carson. He said SDM8 has nearly identical kinetics to UCB-J, while SDM2 appears to enter and leave the brain more rapidly and might be useful for short scans. Both are being tested in human studies, he said. Independently, researchers in Belgium are working on 18F SV2A ligands (Bahri et al., 2017), as are investigators at Invicro, an imaging company based in Boston. Chen predicts the 18F probes will become more widely available in the next year or two, allowing more groups to experiment with the tracer.
UCB-J is being used in small pilot trial of CT1812, a sigma-2 receptor antagonist that purportedly protects against Aβ synaptotoxicity. The 21 patients will undergo PET scans at baseline and after six months’ treatment with either placebo or the drug. Chen said several volunteers have already completed their baseline scans.—Pat McCaffrey and Tom Fagan
Could Better Blood Pressure Management Preserve Cognition?
Is 120 the new 140…for systolic blood pressure, that is? The penultimate day of the Alzheimer's Association International Conference 2018, which ran July 20–26 in Chicago, brought some good news on prevention. Researchers reported data from the SPRINT MIND trial, which tested if reducing systolic blood pressure in older adults to below 120 mmHg staved off cognitive decline. It seems the strategy paid off. Over about three years, on average, 19 percent fewer cases of mild cognitive impairment emerged in the treatment group than in people on standard hypertension therapy, which typically aims for 140 mmHg or less. "This is the first time that a randomized-controlled trial has shown that we can reduce the occurrence of MCI with blood pressure lowering," said Jeff Williamson, Wake Forest School of Medicine, Winston-Salem, North Carolina, a leader of the study.
“This is important work, especially considering the lifestyle- or health-promoting aspects,” said Lon Schneider, University of Southern California, Los Angeles. “It could have profound effects on population incidence and prevention.” Charles DeCarli, University of California, Davis, also praised the trial. “This was a well-designed study with a relatively young age group, ethnic and racial heterogeneity, evidence that blood pressure could be lowered, and which found a moderately large effect, better than with any symptomatic treatment, that’s for certain,” he told Alzforum. “Given that 30–50 percent of the population has hypertension at age 65, this is a big deal.”
The treatment regimen did not reduce dementia incidence, which was the primary outcome measure for SPRINT MIND. Still, clinicians at AAIC did not quibble with that. Schneider, DeCarli, and many others agreed that the trial was probably too short to see an effect on dementia. That stems in part from it being a victim of its own success. The NIH ended the parent Systolic Blood Pressure Intervention Trial (SPRINT) early because reductions in cardiovascular events, including heart attacks, stroke, and all-cause mortality, were so profound. Like SPRINT, the Memory and Cognition in Decreased Hypertension (MIND) sub-study should have run for five years.
As Williamson explained at AAIC, the early termination complicated SPRINT MIND. The trial began recruiting in November 2010, with the last volunteer selected in March 2013. In August 2015 the NIH stopped the trial, unblinding and releasing the data. Concomitantly, investigators stopped dispensing blood pressure medicine to the participants but did continue to offer health advice. This closeout period lasted about a year. About a year after that, investigators restarted follow-up visits to assess longer-term outcomes. Much of the data has yet to be analyzed. The data Williamson presented reflected a mean of 3.26 years of intervention.
SPRINT recruited 9,361 adults at 102 centers throughout the mainland U.S. and Puerto Rico. The average age was 67.9. About 35 percent were women, 30 percent African-American, and 10 percent Hispanic. They had to be older than 50, have a systolic blood pressure (SBP) of 130–180 mmHg, plus one other risk factor for cardiovascular disease. People were excluded if they had prior stroke, diabetes, kidney disease, or dementia.
Investigators randomized 4,683 people to standard care, while 4,678 received intensive management to bring their SBP down to the 120 mmHg goal. At the beginning of the trial, both groups had an average SBP of 140 mmHg. Blood pressure measurements were taken monthly for the first three months, and every three months thereafter. The trial dispensed generic blood pressure medicines that were adjusted at each visit, if necessary, to keep SBP as low as 130 mmHg for the standard care arm, and to 120 mmHg for the intensive treatment.
The pressure lowering worked. Over the first three months, those in the intensive treatment arm saw their SBP fall to almost 120 mmHg. It held fairly steady during the intervention period at about 122 mmHg on average, while those on standard treatment had an average SBP of 135 mmHg. As Williamson and colleagues previously reported, the intervention reduced risk of cardiovascular events, including heart attack, other acute coronary syndromes, stroke, heart failure, and death from cardiovascular disease, by a whopping 27 percent compared to standard care (SPRINT Research Group et al., 2015).
What about cognition? Over the 3.26 years, 175 people in the standard care group and 147 in the intensive treatment group were diagnosed with dementia. That difference was not significant, said Williamson. The difference in MCI incidence was. In standard and intensive arms, respectively, 348 and 285 people were diagnosed with MCI, a 19 percent difference. Incidence of combined probable dementia or MCI was 15 percent lower in the intensive treatment group as well, and that was also significant. These are people who were deemed to have MCI first, and then on a subsequent visit diagnosed with dementia. In SPRINT, MCI was determined by a panel of adjudicators who reviewed data from a SPRINT MIND screening battery, a SPRINT MIND extended cognitive battery, a proxy report to assesses functional decline (a dementia questionnaire), and history of depression and related medications. Adjudicators were trained at the beginning of the trial and recertified yearly. All assessments were recorded and some were sampled later for quality control.
David Knopman, Mayo Clinic, Rochester, Minnesota, chaired a press briefing on the trial but was not involved in the study. He told Alzforum that he considers the methodology for cognitive diagnosis state-of-the-art. “This is the standard way to assign cognitive diagnoses in studies of the scale of SPRINT, where it is simply impossible to have a clinician sit down with each participant and their informant face-to-face,” he told Alzforum.
Rebecca Gottesman from Johns Hopkins University, Baltimore, said the findings were incredibly exciting. “We’ve been looking at this question in various ways, mostly finding that high blood pressure is a risk factor for cognitive decline and dementia, but we have not been able to make the leap that more aggressive lowering of blood pressure would reduce MCI and dementia,” she told Alzforum. “Showing reduced MCI, and MCI/dementia, especially after such a short follow-up, is tremendously important.”
Will this trial prompt a change in clinical practice? Clinicians were not sure. Since the SPRINT trial data came out, a combined American College of Cardiology and American Heart Association panel issued new clinical practice guidelines for managing high blood pressure in adults. The new normal is now 120/80 mmHg or less. Readings between 120/80 mmHg and 129/80 mmHg are deemed elevated, and anything higher than 130/80 mmHg is hypertension (Whelton et al., 2018). In effect, the threshold for hypertension was lowered by 10 mmHg (for a synopsis and a viewpoint see Cifu et al., 2017, and Whelton and Carey, 2017). Because of the new guidelines, the SPRINT MIND findings may not have any radical impact on formal recommendations for treatment, said Knopman. “However, they give primary care physicians more ammunition to recommend aggressive treatment,” he said. Gottesman noted that the cognitive outcomes from SPRINT were not known when the new guidelines were set. “These new [cognition] findings should make an even more compelling case,” she said. “It remains to be seen whether the [cognition data] will motivate people to get their blood pressure under control more than fear of cardiovascular disease would. People are very scared of dementia,” she said.
Despite the enthusiasm, the data may not be generally applicable. Researchers have questioned if people in SPRINT are at higher risk than general population. Julie Schneider, Rush University Medical Center, Chicago, who agreed the findings were good news, added similar words of caution. “We cannot generalize to all persons as these volunteers were selected based on a known history of hypertension treatment, elevated blood pressure, and at least one [cardiovascular] risk factor.” Indeed, researchers led by Paul Munter from the University of Alabama at Birmingham reported that only 16.7 percent of adults in the U.S. who have been treated with hypertension would meet the inclusion criteria. This broke down to 9, 19, and 34 percent of those aged 50–59, 60–74, and 75 or older, respectively (Bress et al., 2016).
Others were concerned about lowering blood pressure in people in their 70s and beyond. Jonathan Schott, University College London, told Alzforum that older adults tend to have stiffer arteries and need a higher blood pressure to maintain perfusion to the brain. Schneider and colleagues recently associated steeper declines in SBP with more brain infarcts in octogenarians (Arvanitakis et al., 2018). “We need more information in these older age groups, especially in those who are accustomed to higher blood pressure and may have pre-existing vessel disease,” first author Zoe Arvanitakis, also from Rush, wrote to Alzforum.
For his part, Schneider advised that any change to blood pressure management be done slowly and cautiously. On a positive note, Williamson and the SPRINT investigators have reported a sub-study of noncognitive outcomes in those aged 75 and older. In short, they seem to tolerate the lower blood pressure, with few reports of falls, hypotension, fainting, or other serious adverse events, while enjoying the same cardiovascular benefits as the younger volunteers (Williamson et al., 2016).
What about Alzheimer’s disease? There’s no evidence from SPRINT that lowering blood pressure has any effect on the main AD pathologies—plaques and tangles. That fits with the current literature. “While there is some limited evidence that hypertension might have an association with tauopathy, the overwhelming evidence to me is that hypertension is a disease/process that impacts later-life cognitive impairment through a cerebrovascular mechanism,” noted Knopman.
Evidence for this emerged in a subgroup of SPRINT patients. Ilya Nasrallah and colleagues at the University of Pennsylvania, Philadelphia, measured changes in brain volume and white-matter lesions using sequential MRI scans. At AAIC, Nasrallah reported no difference in whole-brain volumes between 203 people on standard care and 251 who received the intensive SBP lowering regimen. The latter did have fewer white-matter lesions, however. While the average number of lesions per person grew as the trial progressed, those in the intensive-treatment arm had 18 percent fewer over an average of 47 months follow-up.
Schneider thought the speed of the cognitive and structural brain changes indicate the two may be linked. “The quickness suggests that the hypertension itself contributed to the dementia. Putting that together with the MRI study, then maintaining lower blood pressure reduces white-matter lesions substantially, and flipping that around, those lesions contribute to the expression of cognitive impairment,” he suggested. DeCarli ties this in with AD. “I argue that [controlling hypertension] does not protect from AD, but if you get AD and have brain injury from cardiovascular disease, then you will do more poorly,” he said. “You could hypothesize that if you have AD pathology you would not suffer so drastically if the rest of your body is doing well.”
Where does this go from here? Since mid-life hypertension consistently emerges as a risk factor for late-life dementia, some have suggested a new trial in younger patients. To DeCarli’s mind, that should be the next step. Mortality, cardiovascular disease, dementia—all could benefit from low blood pressure in 50-year-olds, and they can tolerate 120/70 better than 60-year-olds.—Tom Fagan
SPRINT Research Group, Wright JT Jr, Williamson JD, Whelton PK, Snyder JK, Sink KM, Rocco MV, Reboussin DM, Rahman M, Oparil S, Lewis CE, Kimmel PL, Johnson KC, Goff DC Jr, Fine LJ, Cutler JA, Cushman WC, Cheung AK, Ambrosius WT.
A Randomized Trial of Intensive versus Standard Blood-Pressure Control.
N Engl J Med. 2015 Nov 26;373(22):2103-16. Epub 2015 Nov 9
PubMed.
Focused Ultrasound Breaches Blood-Brain Barrier in People with Alzheimer’s
When seduced by microbubbles jiggling to the beat of ultrasonic waves, even the uptight blood-brain barrier lets its guard down. That’s according to the first study to target the barrier noninvasively with focused ultrasound. Researchers used the strategy to temporarily ease open the blood-brain barrier in the prefrontal cortices of five people with Alzheimer’s disease. Led by Kullervo Hynynen and Sandra Black of the Sunnybrook Research Center in Toronto, the scientists propose that this strategy might allow drugs to cross into the brain. The findings, presented at the Alzheimer’s Association International Conference in Chicago July 22–26, were published July 25 in Nature Communications. Studies on animal models presented at the meeting supported the benefit of ultrasound, reporting it allowed experimental therapies, ranging from small molecules to antibodies, to access the brain to clear tau, protect dopaminergic neurons, and boost neurogenesis.
Designing drugs that can transit the blood-brain barrier (BBB) is a major hurdle drug developers struggle to cross. Nearly two decades ago, researchers discovered that low-energy ultrasound waves could temporarily loosen the barrier (Hynynen et al., 2001). Just prior to applying the sound waves, researchers inject microbubbles into the blood. These 1–10 micron diameter chambers of inert gas line the walls of capillaries and larger blood vessels, including those of the BBB. When hit with ultrasound, the microbubbles expand and contract, and this movement loosens the tight junctions between blood vessel cells that cement the barrier. Multiple studies in animals have demonstrated that the ultrasound/microbubble combo temporarily holds the door open for large molecules, such as antibodies, to sneak across (Samiotaki et al., 2015; Nov 2009 conference news). What’s more, researchers have reported that simply opening the barrier has its own benefits, including clearance of Aβ plaques in AD mouse models via activation of microglia (Choi et al., 2008; Burgess et al., 2014; Mar 2015 news).
Does ultrasound open the BBB in people? And is that safe? Those were the questions first author Nir Lipsman and colleagues set out to answer. They enrolled three men and two women with mild to moderate AD who had tested positive for Aβ plaques in the brain on 18F-florbetaben PET scans. They averaged 66 years old. The volunteers received two rounds of focused ultrasound, spaced one month apart. After injecting microbubbles intravenously, the researchers delivered the sound waves in short pulses for up to a minute at an average power of 4.5 watts. This is less than 1 percent of the ultrasound energy used to surgically ablate tissue, such as uterine fibroids or tumors. MRI scans were used to target the sound waves to a small region in the dorsolateral prefrontal cortex. The primary outcome measure of the study was BBB opening, which was measured by tracking diffusion of the MRI contrast agent gadolinium into the brain.
The scientists targeted the waves to a 5mm-by-5mm region for the first round, then doubled the target area for the second round. One patient developed a respiratory infection and was not given the second round of focused ultrasound (FUS), because the researchers did not want to open his BBB during an active infection.
Tiny Door into the Brain. MRI of one patient reveals infiltration of gadolinium contrast in the right frontal lobe immediately after focused ultrasound (middle). The opening is not present before (left) or 24 hours after (right) the ultrasound.
Immediately following the ultrasound, Lipsman observed a discrete infiltration of gadolinium in the targeted region, suggesting disruption of the BBB. One day later, the researchers injected gadolinium again but saw none seep into the brain, suggesting the barrier had resealed. All patients went home the day after the procedure, and none reported any adverse events. According to MRIs, FUS did not trigger hemorrhages or brain swelling, but the researchers did find tiny, round, white-matter hypointensities in the targeted area in two patients immediately following FUS. These hypointensities had disappeared the following day. Lipsman believes they were not microhemorrhages, which would have been likely to persist for more than 24 hours.
The safety findings were welcome news to Cynthia Lemere of Brigham and Women’s Hospital in Boston, who co-chaired the ultrasound session at AAIC. Lemere told Alzforum that a few years ago, the idea of opening the blood-brain barrier in people with AD, who tend to have vascular problems on top of Aβ deposits, sounded risky and counterintuitive. Now this human safety data, together with experiments she and other researchers have done in aged AD mice, are beginning to make the strategy seem promising, she said.
In the small number of people studied thus far, Lipsman and colleagues also looked for changes in amyloid, cognition, and daily functioning. They found no differences between baseline and three months following the second round of ultrasound.
Lipsman told Alzforum that a Phase 2a FUS study is slated to begin in September. It will enroll 30 participants and target larger regions of the brain, including those known to accumulate the most Aβ plaques. He added that if the ultrasound procedure can be established to be safe, then it could potentially be used with any therapeutic that does not easily cross the BBB.
Animal Models Suggest Benefits
Researchers have been studying this approach in different animal models of neurodegeneration. At AAIC, Elisa Konofagou of Columbia University in New York reported that focused ultrasound helped deliver neurotrophic factors into the brain in a mouse model of Parkinson’s disease. First, Konofagou presented findings from the MPTP injection model, in which dopaminergic neurons wither following administration of this neurotoxin. After injecting microbubbles into the blood, she targeted sound waves to the substantia nigra and striatum, the areas most prone to degeneration in PD. She then injected either saline or the neurotrophic protein neurturin, or an adeno-associated virus expressing glial-derived neurotrophic factor (GDNF). Other mice were given these treatments without ultrasound. By the time of treatment, about 40 percent of the dopaminergic neurons in the substantia nigra had died. Konofagou reported that when given together with ultrasound, either growth factor restored about 75 percent of what was lost, and corrected motor deficits the mice had developed by that time.
Konofagou also used the strategy to open a door to the brain for α-synuclein antibodies. She reported that ultrasound significantly enhanced the delivery of the antibodies to the brains of six-month-old A53T and A30T mice, which express mutant forms of human α-synuclein that cause PD. In both cases, the antibodies strongly reduced α-synuclein pathology in animals that had received ultrasound to open the BBB.
Previously, researchers led by Hynynen and Isabelle Aubert at the Sunnybrook Research Institute in Toronto reported that in the TgCRND8 mouse model of amyloidosis, ultrasound alone activated microglia, stimulating them to remove Aβ plaques (Jordão et al., 2013). However, Konofagou told Alzforum that ultrasound neither activated microglia nor reduced α-synuclein deposits in the PD models. Perhaps microglia are primed differently in the presence of amyloid, she speculated. Jürgen Götz of the University of Queensland in Brisbane agreed. He added that extracellular Aβ plaques—as opposed to intracellular α-synuclein inclusions—might be more amenable to clearance by activated microglia.
Götz had previously reported that merely opening the blood-brain barrier—sans therapeutics—motivated microglia to mop up Aβ plaques, in this case in APP23 mice (Mar 2015 news). Götz used scanning ultrasound, in which the ultrasound waves are delivered in short pulses throughout the entire brain. However, this was less efficient in clearing tau pathology, he found. It did aid the delivery of RN2N, a single-chain, tau-specific antibody fragment, into the brains of P301L mice, where the fragment made its way into neurons and dendrites. The combination treatment reduced phospho-tau more than the antibody alone did (Nisbet et al., 2017).
At AAIC, Götz extended these findings to a model of frontotemporal dementia with parkinsonism. K3 mice express the K369I mutant of tau in the substantia nigra. They rapidly develop neurofibrillary tangle pathology and an early onset parkinsonian motor phenotype, and anti-tau antibodies reduce their phospho-tau and tangles but not their motor problems (Ittner et al., 2008; Ittner et al., 2015). At AAIC, Götz showed that if the BBB was repeatedly opened with weekly scanning ultrasound beginning at five weeks of age, not only did the mice accumulate less phospho-tau and tau tangles than controls, but their motor coordination, as tested by the Rotarod test, improved after the fifth sonication. This suggested that when given enough times, scanning ultrasound alone is beneficial.
“We have done multiple studies with these mice in the past, and it was almost impossible to see improvement in motor symptoms with an antibody alone,” he told Alzforum. “Now, using ultrasound alone, we finally see real improvement.”
Taking a different tack, Aubert has used focused ultrasound to promote adult neurogenesis and neuronal survival, which wane as AD progresses. Previously, Aubert and colleagues reported that temporarily easing the BBB with focused ultrasound stimulated adult neurogenesis in the hippocampus (Scarcelli et al., 2014; Mooney et al., 2016). At AAIC, she reported that adding a pro-survival molecule into the mix boosted neurogenesis further. Specifically, Aubert treated TgCRND8 mice with ultrasound alone or in combination with a small molecule agonist of tropomyosin receptor kinase A (TrkA), a high-affinity receptor for neurotrophin. Ultrasound alone triggered pro-survival responses, such as MAP kinase activation, but adding the TrkA agonist dramatically enhanced them. The same was true in non-transgenic mice, suggesting that the beneficial effects of the agonist were not dependent on Aβ pathology.
Aubert has also used ultrasound to deliver intravenous immunoglobulins (IVIg) into the brains of TgCRND8 mice. IVIg, a mixture of antibodies pooled from blood donors, has been tested as a potential therapy for AD but failed in Phase 3 (May 2013 news). The rationale was that anti-Aβ and other antibodies might promote beneficial immune responses, clear Aβ, and restore brain function. At AAIC, Aubert reported that FUS facilitated the delivery of immunoglobulin from the mixture into the brains of TgCRND8 mice; alas, 20 days later, the researchers were surprised to find that the FUS had not promoted more clearance of Aβ than did IVIg alone, but rather had enhanced neurogenesis.
Aubert proposed that cytokine changes brought about by both the ultrasound and IVIg somehow influenced the birth rate of newborn neurons. As to which cells are expressing those cytokines, Aubert reported activation and proliferation of microglia in response to ultrasound. Lemere pointed out that the complement pathway, which is upregulated in activated glial and myeloid cells, might play a role in promoting neurogenesis in response to ultrasound. She added that it may also be possible that circulating monocytes gain access to the brain while the BBB is open. Konofagou agreed that infiltration of monocytes was a possibility. Both researchers agreed that if, and how, peripheral cells mediate ultrasound effects need further study.—Jessica Shugart
Four Immunotherapies Now Banish Amyloid From the Brain
After years of fits and starts, anti-amyloid immunotherapies are finally hitting their target effectively. At least four drugs have now demonstrated the ability to clear plaques from the brain: aducanumab, gantenerumab, Lilly’s LY3002813, and BAN2401 (Jul 2018 conference news). At the Alzheimer’s Association International Conference, held July 22–26 in Chicago, researchers presented new data from gantenerumab and LY3002813, aka N3pG/donanemab. It clinched the case that these antibodies can mop up brain amyloid, bringing many people with early symptomatic Alzheimer’s disease below the threshold for amyloid positivity. At one to two years, this clearance took a long time. But still: Gregory Klein at Roche claimed that two years of treatment with high-dose gantenerumab essentially resets a person’s trajectory of amyloid accumulation. “We are setting back the clock by 15 years,” Klein told the audience.
To achieve these rates of clearance, researchers have had to greatly boost antibody dose, in many cases quadrupling the amounts used in earlier, unsuccessful trials. The same was done for two additional anti-amyloid antibodies that did not report on amyloid removal at AAIC. Crenezumab’s dose was ramped up midstream in the Colombian ADAD trial, and is high in the ongoing CREAD Phase 3 program in LOAD; solanezumab’s dose was pushed up in DIAN and A4, two ongoing secondary prevention trials (Jun 2017 news).
These high doses bring a greater risk of infusion site reactions and ARIA-E, the occurrence of leaky blood vessels causing edema in the brain. In Chicago, scientists argued that these side effects are manageable with careful monitoring of patients. Moreover, ARIA-E can be lessened by gradually titrating up the antibody dose, they said.
Other researchers found the data encouraging. “It’s impressive there are now four drugs that can remove amyloid plaques from the brain,” Randall Bateman of Washington University, St. Louis, wrote to Alzforum. However, clinicians noted that the jury is still out on how much this will help AD patients. “Several of the antibodies are looking good at removing amyloid, but the clinical efficacy still needs to be demonstrated,” said Ron Petersen of the Mayo Clinic in Rochester, Minnesota.
Brain Drain.
Amyloid scans from three patients on gantenerumab at baseline (top row), one year (middle), and two years (bottom). Reduction shown in centiloids. [Courtesy of Greg Klein.]
Previously, at the 2017 CTAD conference, Roche scientists presented six- to nine-month gantenerumab data from ongoing open-label extensions of two Phase 3 studies. Participants were titrated up from 105 or 225 mg subcutaneous gantenerumab to 1,200 mg, and after nine months, their amyloid had receded by 15 percent, with one-third of participants dipping below the threshold of brain-wide amyloid positivity (Dec 2017 conference news).
In Chicago, Klein added data from the one- and two-year timepoints. The researchers transformed SUVR PET data to the centiloid scale, following a new convention that eases comparisons between data obtained with different tracers (Feb 2013 conference news; Nov 2014 news). At the one-year mark, 61 participants had amyloid PET scans; 43 of them had received at least six doses of 900 or 1,200 mg. Overall, their amyloid reduction was still similar to that at nine months, Klein showed. The 15 participants from the Scarlet RoAD extension notched drops of 24 centiloid at nine months and 29 at one year. The 46 participants from Marguerite RoAD stayed steady at about 45 centiloid reduction at both timepoints. (Why the difference? Scarlet RoAD enrolled prodromal AD at baseline compared with mild to moderate AD for Marguerite RoAD. The former had less amyloid at baseline, and had received fewer high doses of gantenerumab by one year, Klein explained.)
By two years, however, things changed dramatically. The 28 participants who underwent PET at this timepoint on average had received 18 to 20 doses of 900 mg or higher. Scarlet RoAD participants lost 46 centiloids worth of amyloid, while Marguerite RoAD participants who had started out on placebo and switched to gantenerumab lost a whopping 78 centiloids. Participants previously in the active arm of Marguerite RoAD saw their scans shrink by 48, because this group had little amyloid left to lose, Klein noted. Lilly’s Adam Fleisher confirmed that with his company’s new antibody, too, the higher a person’s baseline SUVR, the more amyloid reduction they tended to post (see below).
For all groups, baseline centiloid averaged 80, and the reduction after two years was 59. A centiloid of 24, corresponding to an SUVR of 1.4, is the threshold for amyloid positivity on these florbetapir scans, Klein said. By the two-year mark, half the cohort had fallen below it, and the remainder were on a trajectory toward the amyloid floor of negative 50 centiloids, Klein said. Amyloid declined fastest in people with heavy loads, and more slowly as people approached the floor, he added.
But will this help people with their cognition and function? “We are quite optimistic that these large reductions will correspond to a clinical benefit,” Klein said.
And how safe are these high doses? In Chicago, Roche’s Danielle Abi-Saab presented safety findings from the Marguerite RoAD extension of 219 participants, and Mirjana Andjelkovic from the Scarlet RoAD extension, with 154 participants. In both, the higher doses brought more ARIA-E. A third of participants in the Marguerite RoAD extension developed it, compared with 12 percent in the original study. In the Scarlet RoAD extension, it was 28 percent. APOE4 carriers had the highest rates, topping out at 38 percent in the Marguerite RoAD extension. Without titration, 58 percent of APOE4 carriers would have been expected to develop ARIA-E at these doses, Abi-Saab noted, suggesting that titration worked to lower incidence.
The majority of ARIA-E instances were detectable only on MRI scans. That said, one-quarter of participants with ARIA-E in either cohort did report symptoms, typically headache. Four participants in the Marguerite RoAD extension, and three in Scarlet RoAD, had more serious reactions, including seizures, confusion, ischemic stroke, and neurological symptoms on one side of their body. After discontinuing drug, all these conditions cleared up, the speakers said. Everyone with ARIA-E had an MRI scan per month, and on average, ARIA-E resolved fully over three months. The majority of participants with ARIA-E were able to continue in the study.
The researchers saw fewer instances of ARIA-H, or microhemorrhages. These occurred in 9 percent of Marguerite RoAD, and 6.5 percent of Scarlet RoAD extension participants. The protocol forced an end to treatment in the majority of these participants.
In both studies, about one-third of participants developed rashes at the infusion site. These were typically mild, though one person dropped out due to this reaction.
At high doses of gantenerumab, safety remains favorable, and ARIA events are manageable, the speakers concluded. Data from these extension studies were used to design the Phase 3 GRADUATE 1 and 2 studies of subcutaneous gantenerumab titrated up to high doses. Both trials have started enrolling.
Lilly’s Anti-Pyroglutamate Aβ Antibody Removes Plaque Quickly
The findings for Eli Lilly’s N3pG are similar. This newer antibody targets a pyroglutamate form of Aβ that is particularly prone to aggregate and is found mostly in plaques. As with other antibodies, Lilly’s initial dosing turned out to be too timid. In Phase 1, only the highest dose used, 10 mg/kg, produced meaningful brain exposure. At this dose, brain amyloid dropped by about 44 centiloid, with almost no ARIA. But there was a catch. Most participants made anti-drug antibodies, and six of 37 had an infusion reaction (Aug 2016 conference news).
Fast Removal.
Amyloid scans from three patients taking N3pG. Baseline (top row), three months (middle), and six months (bottom). Readings given in SUVR/centiloids; red numbers are below cutoff for positivity. [Courtesy of Adam Fleisher.]
This prompted Lilly to explore safety and immunogenicity of higher doses, and in Chicago, Fleisher presented interim data from this ongoing Phase 1b study. The 58 participants have prodromal to moderate AD, and range around 73 years old with an MMSE score of 21. About three-fourths carry an APOE4 allele, and their average baseline centiloid was 104.
The study tests three dosing regimens. In one, participants receive a single dose of 10, 20, or 40 mg/kg. In the second, they get 10 mg/kg every other week for 24 weeks. The third is a chronic dosing schedule of 10 or 20 mg/kg every month for 16 months, or until the participant’s amyloid scan turns negative. Each dosing scheme includes nine to 12 participants randomized 3:1 to drug or placebo, except for the 40 mg/kg single dose, which has only two participants. Besides regular assessment for anti-drug antibodies, amyloid and MRI scans happen quarterly, and cognitive testing every six months.
Fleisher reported to the AAIC audience that plaque load drops after even a single dose of the antibody, and stabilizes at the new level during follow-up. Higher doses effect a greater reduction, with the highest so far being an average drop of 70 centiloid with six months of 20 mg/kg. In this highest dose group, three of six patients have become amyloid-negative so far, Fleisher said. He added that not everyone responded to 10 mg/kg chronic dosing, but all responded to 20 mg/kg.
Alas, Lilly also saw more ARIA at higher doses. About one-quarter of dosed participants developed ARIA-E. Two people reported symptoms such as headaches, confusion, and sleepiness, which went away after dosing stopped. In contrast to the first trial, just one participant experienced a rash at the infusion site, and only on the first dose. As before, however, nearly all patients made anti-drug antibodies. These have caused no apparent side effects thus far. They did not affect Np3G’s efficacy, either, except in one patient whose B cells pumped out massive amounts of antibody, more than 100,000 per drug molecule. Researchers stopped treatment in this participant, as well as in another with a microhemorrhage and in 11 with ARIA-E. Lilly is closely monitoring all participants for 18 months.
While this study is ongoing, Lilly has advanced N3pG to Phase 2, where it is being tested in combination with Lilly’s BACE inhibitor LY3202626. Dubbed TRAILBLAZER, this trial is the first to test a combination of disease-modifying therapies for AD, Lilly’s Michael Irizarry said at AAIC. The trial is currently enrolling, targeting 375 participants with early symptomatic AD. They will be divided evenly among three arms. One arm will receive placebo, another N3pG, and the third both N3pG and the BACE inhibitor. The primary endpoint will be the integrated Alzheimer’s Disease Rating Scale (iADRS) developed by Lilly, with ADAS-Cog, CDR, MMSE, and ADCS iADL as secondary outcomes. The researchers will also look at volumetric MRI, amyloid PET, and tau PET, as well as safety and tolerability.
Notably, the trial will enroll only people whose neurofibrillary tangle buildup falls within a low-to-medium range defined as an SUVR of 1.15 to 1.36 by flortaucipir PET. This is because people with less tau than the lower cutoff show little cognitive decline over one to two years, while people with more tau than the upper cutoff might be at too advanced a stage to still benefit from an anti-amyloid treatment. Stratifying participants in previous trials by their tau PET signal has shown a close correspondence between tau pathology at baseline and subsequent rate of cognitive decline, Irizarry noted.—Madolyn Bowman Rogers.
Memory Slips as Soon as Amyloid Appears, Two Decades Before Dementia
How early in Alzheimer’s disease does a person’s memory falter? As soon as amyloid starts to build up, according to speakers at the Alzheimer’s Association International Conference in Chicago July 22–26. Even before an amyloid scan turns officially positive, subthreshold accumulation correlates with subtle memory deficits and presages future decline. Researchers running the A4 secondary prevention trial of late-onset AD noted that cognitive deficits are already detectable at baseline in this otherwise preclinical population. Swedish researchers attempted to quantify a rate of decline at the preclinical stage, to estimate what size trials will be needed to detect drug effects at this time point.
“This is a tremendously exciting time in AD research. Our concept of Alzheimer’s has evolved, and we now think of it as a pathophysiological continuum,” said Reisa Sperling of Brigham and Women’s Hospital, Boston. She believes the ability to detect initial cognitive changes opens the door for ever earlier-stage secondary and even primary prevention studies that are now gearing up across the field.
The Swedish group also presented an updated, detailed timeline of biomarker change in sporadic AD. As predicted, amyloid builds up long before tau neurofibrillary pathology and then cognitive change. For both amyloid and tau, cerebrospinal fluid biomarkers change up to a decade before the corresponding PET signal rises. These CSF markers precede a dementia diagnosis by 30 years, while cognitive change becomes detectable 20 years before dementia. The data reinforce the idea that Alzheimer’s is a disease that occurs over decades.
Colin Masters of the University of Melbourne, Australia, noted that data on longitudinal CSF and PET changes are beginning to converge across all the major observational studies. “We are on a steep learning curve when it comes to defining the lower thresholds and their cut points,” he wrote to Alzforum. “We are now seeing the limits of sensitivity and specificity of the various PET/CSF technologies.” The ability to detect these very small changes may further refine concepts of disease, he suggested.
CSF and PET Separate.
Preliminary estimates from longitudinal ADNI data suggest refinements to the staging curves, with CSF markers preceding PET signal by many years. This analysis uses time to brain-wide amyloid positivity as the reference point. [Courtesy of Philip Insel and Niklas Mattsson.]
The first evidence tying subthreshold levels of brain amyloid to worse cognition came earlier this year, when William Jagust and colleagues at the University of California, Berkeley, reported that amyloid-negative ADNI participants who were accumulating plaque scored more poorly than non-accumulators on memory tests over time (May 2018 news).
In Chicago, Denise Park of the University of Texas, Dallas, corroborated this finding with data from the Dallas Lifespan Brain Study. This observational cohort study launched in 2008; every four years it examines 500 healthy and cognitively normal adults ranging from age 20 to 89, who were amyloid-negative at baseline. On average, it takes 10 or more years for plaque growth to reach the brain-wide threshold for amyloid positivity.
The news here is that cognitive effects became apparent before the positivity threshold. In the Dallas cohort, people who were accumulating subthreshold amyloid, particularly in posterior brain regions such as the precuneus and posterior cingulate, scored worse on memory tests at their first follow-up than at baseline. Executive function and other cognitive abilities remained stable (Kennedy et al., 2012; Bischof et al., 2016; Farrell et al., 2017; Farrell et al., 2018, paper in press).
Other studies have linked subjective memory complaints to elevated amyloid, even when people still score within the normal range on standard cognitive tests (Amariglio et al., 2012). The new findings show that these subjective memory complaints can be measured and tracked, Park noted. The data further strengthen the idea that any amyloid accumulation indicates a person is on the path to AD. “Amyloid foretells an individual’s future,” Park said.
Sperling reported complementary findings from A4. The trial is fully enrolled, with 1,169 participants between ages 65 and 85 being seen at 67 sites across the United States, Canada, Australia, and Japan. All participants are cognitively normal, with a CDR of zero and an MMSE score between 25 and 30. All have a positive amyloid scan, with an average SUVR of 1.33.
To find these participants, A4 researchers screened 4,486 people by amyloid PET. One-third were amyloid-positive and, importantly, compared with their amyloid-negative peers, they scored worse on the PACC, a cognitive composite designed to pick up the earliest signs of cognitive change (Jun 2014 news). The difference in scores was small, but highly statistically significant, Sperling noted. Likewise, the amyloid-positive group scored worse than amyloid-negatives on the Cognitive Function Index (CFI), a questionnaire that assesses subtle functional deficits (Mar 2015 news). As with the PACC, the difference was small but robust, with a p value of below 0.0001.
Joshua Grill of the University of California, Irvine, who studies disclosure of brain amyloid status in A4, showed data suggesting that even at baseline, people who are accumulating amyloid appear to be aware at some level that their memory has changed. Before participants learned their amyloid status, the amyloid-positive group scored slightly higher on a measure of anxiety than amyloid-negatives, and also self-reported more concerns about having Alzheimer’s.
Overall, these A4 data suggest it will be possible to detect a slowing of decline in people at preclinical stages of AD, Sperling said.
Preclinical Cognitive Decline.
Around the world, three cohorts converge to find that amyloid-positive people (red) take six years to decline from healthy control levels (black) to a marked memory deficit (purple line) on the PACC cognitive composite. [Courtesy of Oskar Hansson.]
Oskar Hansson of Lund University, Sweden, also spoke to this point. He combined six years of findings from three longitudinal cohorts—the North American ADNI, the Australian AIBL, and the Swedish Biofinder Study—to estimate the degree of memory decline in cognitively normal people with elevated brain amyloid. The three studies comprised 350 people with elevated amyloid, and 770 who were amyloid-negative. Demographic factors such as age and education varied widely between cohorts, as did study protocols, recruitment criteria, and cognitive testing.
Despite these differences, all three studies recorded a similar magnitude of decline in amyloid-positive people, amounting to about a 0.5 point deficit in PACC score over four years. Amyloid-negative participants, by contrast, remained stable. At this rate of decline, a four-year preclinical study enrolling 500 participants per arm would have 80 percent power to detect a 50 percent slowing of cognitive decline, Hansson estimated. At 800 participants per arm, a trial could detect 40 percent slowing. Large studies with longitudinal follow-up could see subtle change in cohorts that still score in the normal range on cognitive tests, Hansson concluded.
John Breitner of McGill University in Montreal challenged this conclusion, noting that a slowing of 20 to 30 percent is more realistic for most drugs. The estimates suggest that a trial in this preclinical population would need to enroll thousands of participants, Breitner said. “It looks daunting,” Breitner added, but Hansson pointed out that longer studies would have more power and could bring these numbers down. Hansson told Alzforum that a four-year study in this population would need 1,700 participants per arm to detect a 25 percent slowing of decline.
In addition, it was unclear how many of the amyloid-negative group in these three cohorts were in fact accumulating sub-threshold plaque. Because accumulators already have some cognitive decline, their presence in the control group could be lowering the power to detect an effect, noted Suzanne Hendrix of Pentara Corporation, Salt Lake City. Hansson agreed that future studies should use non-accumulators as a control.
Despite the evidence linking amyloid accumulation to cognitive decline, are plaques directly responsible for this decline? A wealth of imaging data now shows that where amyloid goes, tau tangles follow, and these correlate more closely with cognitive loss. Niklas Mattsson, also at Lund, added more data in favor of this sequence of events. Such a sequence is well-established in dominantly inherited AD, where researchers have been able to assemble a more detailed timeline of biomarker changes because they can pinpoint estimated onset age (Jul 2012 news). This has not been possible in sporadic AD preclinical cohorts, where a given person’s onset age is unknown.
To perform a similar analysis in sporadic disease, Mattsson and Philip Insel at Lund analyzed ADNI data from 43 amyloid-negative, cognitively normal controls, 34 amyloid-positive controls, 35 people with MCI due to AD, and 13 AD patients. Each participant had undergone an average of three amyloid PET scans over the course of about five years, and the researchers used these serial scans to calculate each person’s rate of amyloid accumulation. Then they applied this rate to estimate when each person would reach the threshold for amyloid positivity. For people who were already positive, the researchers regressed their accumulation rate to find the likely date when they had first become positive. The time of PET positivity was taken as year zero. This time point allowed the researchers to compare other biomarker findings and estimate when each became abnormal. Abnormality was defined as diverging more than two standard errors away from the level in amyloid-negative controls.
Using this method, Mattsson and colleagues found that CSF Aβ42 first starts dropping more than 12 years before PET scans become positive. CSF total tau starts rising around the same time, with p-tau following two years after. In other words, fluid biomarkers moved long before PET scans detected measurable protein accumulation. These data reinforce a growing realization in the field that CSF and PET measure different aspects of disease (Aug 2017 conference news). Masters noted that the relationship between tau phosphorylation and tangle formation remains murky. Some phosphorylation sites, such as Ser202/Thr205, define a pre-tangle structural change. “That structure has not yet been evaluated in relation to the various tau PET ligands. It may turn out that the CSF p-tau signal is quite different from the tau PET signal,” he said.
In the ADNI cohort, a tau PET signal in the medial temporal lobe came up about five years before brain-wide amyloid positivity. Mattsson told Alzforum that this signal was more widespread than normal age-related tau accumulation, which tends to be largely confined to the entorhinal cortex. Within a couple of years more, a tau signal became detectable in the medial parietal lobe, as well. The findings indicate that numerous biomarker changes occur before a positive amyloid PET scan flags a person as having AD. However, Mattsson noted that tau load remains low at these early stages, and does not reach the levels seen in MCI until about a decade after a positive amyloid PET scan.
Cognition changed last in this ADNI sample. Logical memory scores trended down right around the time of amyloid positivity, PACC scores followed a couple of years later, and MMSE scores waned shortly after that. Thus, cognitive change followed closely after the first signs of tau tangles spreading, in agreement with other research linking tangles to cognitive decline (Aug 2017 conference news).
Mattsson cautioned that the sample was small and these estimates are rough (see image above). More longitudinal data from larger studies will be needed to make them precise. However, the findings to date largely agree with those from the Australian Imaging, Biomarker, and Lifestyle (AIBL) Flagship Study of Ageing, which has found about a 20-year timeframe between amyloid positivity and mild cognitive impairment in sporadic disease (Dec 2014 conference news).
In sum, Mattsson’s analysis adds independent confirmation that memory decline can already be detected about 25 years before dementia onset. The data set suggests that about 35 years elapse between the first biomarker changes, at least as currently detectable, and an AD diagnosis, Mattsson said.—Madolyn Bowman Rogers
Surprise: HDAC Goes Down, Not Up, in Alzheimer’s Disease
As gene regulation changes in Alzheimer’s disease, scientists believe that histone deacetylases (HDACs) go into overdrive, shutting down transcription of certain genes. Consequently, several research groups are exploring the potential of HDAC inhibitors as AD therapeutics, with at least two trials currently enrolling. The recent development of a PET tracer that recognizes class I HDACs in the brains of living people provides a valuable tool for such trials. Now, however, the first HDAC PET data from people with AD upends previous findings. At the Alzheimer’s Association International Conference in Chicago July 22–26, Tharick Pascoal of McGill University, Montreal, reported that HDAC levels drop as disease advances.
“That was a surprise to us. We expected the opposite result,” Pascoal told Alzforum. At first, the researchers worried that there might be errors in their methodology; however, validation by a second group studying an independent cohort convinced them the finding was real, and robust. “I’ve never seen two independent PET studies where the images were so similar,” Pascoal said.
The HDAC ligand, [11C]Martinostat, was developed by Jacob Hooker of Massachusetts General Hospital, Boston, in collaboration with other groups. The tracer enters the brain readily and binds the three class I histone deacetylases, HDAC1, 2, and 3 (Wang et al., 2014; Wey et al., 2015). In healthy young controls, the tracer lights up specific brain regions, with the strongest signal in the putamen and cerebellum and weaker signal in the hippocampus and amygdala. The same pattern occurs in all volunteers, suggesting tight regulation of HDAC expression (Aug 2016 news).
High AD Pathology, Low HDAC. In AD brains, amyloid plaques (left) and tau tangles (middle) accumulate while HDAC levels (right) wane in posterior cingulate cortices (arrow), compared with healthy elderly control brains. [Courtesy of Tharick Pascoal.]
Pascoal, working with Pedro Rosa-Neto and Serge Gauthier at McGill, used the tracer to scan 48 volunteers seen at McGill. Two of them were young healthy controls, 15 were cognitively healthy elderly, 15 had MCI, and 16 were diagnosed with AD. All also underwent amyloid scans with AZD4694, aka NAV4694 (May 2010 news), and tau imaging with MK6240. The cognitively healthy older participants were amyloid-negative.
In Chicago, Pascoal reported that brain regions that were positive for both plaques and tangles took up less Martinostat signal than control brains did. The affected regions— posterior cingulate cortex, precuneus, hippocampus, inferior parietal lobe, and lateral temporal lobe—all form part of the posterior default mode network, which accumulates pathology early in Alzheimer’s disease. The greater the AD pathology in these regions was in a person, the lower his or her Martinostat signal. The difference between cognitively healthy elderly and AD brains was stark, with almost no overlap between the two. In terms of SUVR, the Martinostat signal in young healthy controls was 1.5; in healthy elderly it ranged between 1.5 and 1.3; and in AD it ranged from just under 1.3 to 1. People with MCI had an intermediate level of Martinostat signal, suggesting that levels drop as disease develops, Pascoal said.
Pascoal and colleagues compared their data to a separate study done by Hooker and colleagues at the Martinos Center for Biomedical Imaging at MGH on 23 young people, 13 cognitively healthy elderly, and 10 AD patients. The two research groups used different protocols and analyses; even so, they saw the exact same HDAC pattern. “That gives the findings a lot of rigor,” Hooker told Alzforum. At AAIC, Pascoal presented pooled analyses from both cohorts.
What do low levels of HDAC in AD mean? Hooker noted the reduction in HDAC either could be compensatory or it could be part of disease pathology. A statistical analysis suggested the latter was more likely. Pascoal correlated regional HDAC, amyloid, and tau pathology to MMSE scores. He found that adding HDAC levels to the model increased its power to predict cognitive deficits. Amyloid and tau together explained about 70 percent of the sample’s variance in cognition, and with HDAC added, this rose to 90 percent. The data suggest that HDAC levels may mediate some of the damaging effects of Alzheimer’s pathology on cognition, Pascoal said.
Why has previous human research not seen this dramatic decrease in HDAC? Pascoal suggested this is because previous research was all postmortem and pathologists looked in the wrong regions. Most previous studies examined the medial temporal lobe or prefrontal cortex, where there is no change in Martinostat signal. The McGill group followed up on their own live imaging findings with a western blot analysis of postmortem samples from nine healthy elderly and six AD brains. That confirmed low levels of HDACs 1, 2, and 3 in AD brains in those same posterior DMN regions that had low Martinostat signal in the imaging study. “The take-home message from this research is that you have to be attentive to brain region,” Rosa-Neto told Alzforum. “The disease process is dynamic, and Alzheimer’s pathophysiology varies from region to region.”
In addition, much of the previous research on HDACs has been done in mouse models, which do not always reflect human biology. Several studies have reported less histone acetylation in the hippocampi of various neurodegenerative disease models, including APP/PS1 AD mice, than in wild-type, and HDAC inhibitors improved learning in these models (Jun 2004 news; May 2007 news; Francis et al., 2009; Kilgore et al., 2010; Mar 2012 news).
For their part, Pascoal and colleagues performed a western blot analysis of postmortem samples from two rat models of AD. The R-Thy1-APP line (Leon et al., 2010) carries a human amyloid transgene and does not develop neurofibrillary tangles; it showed no change in class I HDAC levels compared to wild-type rats of the same age, Pascoal said in Chicago. On the other hand, the TgF344 rat (Cohen et al., 2013) develops both amyloid plaques and tau tangles; it had lower HDAC1 and 2 levels than wild-type. The findings suggest that, just as in human brain regions, both amyloid and tau pathology are required to change HDAC levels, Pascoal said. Both findings contrast with mouse data showing increased HDAC in AD models, suggesting the mouse findings might not translate to other species.
There is another wrinkle, though. The Martinostat tracer does not distinguish between particular class I isoforms. Li-Huei Tsai of the Massachusetts Institute of Technology noted that some of these may move in different directions in AD. For example, Tsai has evidence that HDAC2 rises in the hippocampi of AD mice, whereas other isoforms, such as HDAC1, may fall. HDAC2 has been tied to harmful effects, while HDAC1 and a class III isoform, SIRT1, seem to be neuroprotective in mice (Dec 2008 conference news; May 2009 news; Feb 2010 conference news). “Concerning using HDAC inhibitors for treatment, obviously isoform specificity would be very important,” Tsai wrote to Alzforum. She did not hear Pascoal’s AAIC talk.
Hooker acknowledges that Martinostat’s lack of specificity is a limitation. Alas, he noted, available drugs don’t distinguish between class I isoforms, either. “We believe the pan-class Martinostat is useful to judge the overall HDAC, or epigenetic, ‘tone,’” Hooker told Alzforum. Another potential limitation of the tracer is that it measures HDAC protein level, not necessarily its activation state. “We assume that HDAC density and activity are linked, but HDAC [regulation] is complicated. As with all PET imaging agents in humans, we have to be cautious with the exact interpretation,” Hooker noted.
How does this information affect HDAC inhibitor trials? The McGill group recommended caution. “Because HDAC class I levels are already low in AD, there is the potential that reducing them further could cause harm,” Pascoal said.
A Phase 1 open-label dose-finding study of the HDAC inhibitor vorinostat is currently enrolling in Germany. Vorinostat inhibits class I and class II HDACs. The McGill group has shared their data with the German investigators so they can take it into account. Notably, the class I HDAC inhibitor valproate was previously found ineffective for managing neuropsychiatric symptoms in AD, and it also associated with worse cognitive decline (Fleisher et al., 2011).
Meanwhile, researchers at the University of California, Irvine, are investigating the class III HDAC inhibitor nicotinamide for AD. They wrapped up a Phase 1 safety study of 50 AD patients in 2014, and are currently enrolling for Phase 2. The Martinostat findings provide no information on class III HDACs, Pascoal noted.—Madolyn Bowman Rogers
Weeklong Chinese Challenge Reveals Subtle Memory Problems
Could learning Chinese make a game out of cognitive testing—all the while spotting earlier stages of decline within a week’s time? Perhaps, according to work presented at the Alzheimer’s Association International Conference, held July 20–26 in Chicago. When Jenalle Baker of the Florey Institute, Melbourne, Australia, challenged older adults to memorize the English equivalents of 50 Chinese characters, within one week she started to detect subtle learning problems tied to brain amyloid deposition that would take a year to discern with traditional tests. Also at the meeting, other researchers showed off how smartphone and digital pen technology can simplify and improve cognitive testing aimed at detecting the smallest of dips in mental function.
With the growing use of amyloid PET scanning, researchers discovered that as people accumulate amyloid, their cognitive power wanes long before dementia appears (Feb 2018 news; Baker et al., 2017; Aug 2018 conference news). But the decrements are small, and take more time to detect than is practical for clinical trials. That is a serious problem for early stage therapeutic studies, which need faster cognitive outcome measures.
For example, Baker, who works with Yen Ying Lim at Florey Institute, showed data from the longitudinal Australian Imaging, Biomarker & Lifestyle Study of Ageing (AIBL). In that cohort, amyloid-positive and -negative, cognitively normal people start out performing the same on a learning and memory task. The test asked participants to learn and recall the locations of abstract shapes on a grid, a measure of paired association learning that is sensitive to the disruption of hippocampal function in early AD. In a single session, both amyloid-positive and -negative people learned quickly and got better with practice, improving their scores equally on repeated trials given on a single day. But in the following years, the groups diverged: Amyloid-negative people continued to improve on the test, while scores for amyloid-positive people stayed flat. Clearly, the amyloid-positive people were worse at acquiring new information. Baker’s problem? It took a year for the difference between the groups to become significant.
Baker wondered if she could devise a test to pick up that difference sooner. She developed the Online Repeated Cognitive Assessment (ORCA), a more challenging version of paired association. ORCA asks volunteers to learn to match 50 Chinese characters with their meaning, spoken in English. People did this at home, via the Internet, 25 minutes per day for six days. The outcome was the number of correct and incorrect guesses. All data went to a central server for analysis.
Baker evaluated the test in 30 amyloid-negative and 20 amyloid-positive, cognitively normal AIBL participants. Both groups were equal in average age, hippocampal volume, and scores on established cognitive tests.
Over the six-day testing period, all participants learned to associate the words with the characters, but the amyloid-positive group made significantly more mistakes than their amyloid-negative counterparts. The difference emerged by the second day of testing, and grew in succeeding days. By day six, the magnitude of the impairment was large, with the amyloid-positive group showing an effect size of 1.69 relative to the amyloid-negative group. That is a far greater and faster difference than traditional pen-and-paper measures, where effect sizes even after a year stay within 0.15 to 0.3 (Baker et al., 2017). “ORCA takes one week, not one year,” Baker said.
The result needs to be repeated in larger and independent samples, but Baker thinks the data look promising. ORCA may be suitable for widespread screening for clinical trial candidates, or to triage people for diagnostic follow-up. Baker said preliminary results with this initial sample of 50 people suggest that the test can discriminate amyloid positivity with 70–80 percent accuracy and a high test-retest reliability of 0.9. “We want to move toward establishing a cutoff score, but need to test more participants first,” she said. With more data, ORCA could also be used to evaluate treatment effects, she suggested.
Baker said it will be important to determine whether a person’s performance on ORCA is influenced by cognitive enhancers or other medications older people take, or by sleep quality, or genetic variants such as ApoE4.
One audience member questioned how the researchers keep people engaged in this regimen, which would seem to take considerable effort on the part of participants. Lim replied that, au contraire, participants reported enjoying taking this test. She believes this is because the test does not rely on abstract symbols. “It has a real-world aspect that drives engagement,” Lim said. One participant told Baker that, while walking in Melbourne’s Chinese district, he recognized the character for noodle on a restaurant sign. “People feel like they are learning something,” Baker said.
This format is only useful in English speakers who know no Chinese, which rules out readers of Japanese, as well, as the language uses many of the same pictographs. Lim told Alzforum her group is trying to develop tests with other pairings, such as birds and their songs.
The test is available online, and a mobile phone app is under development, Lim said, adding that interested researchers should contact her.
Reality Check. Top graph shows tidy, but imaginary, results of cognitive testing of amyloid-positive (black) and –negative (blue) adults, versus the bottom graph of real-world data. Could a smart phone app help clean up the mess? [Courtesy of Jason Hassenstab.]
Jason Hassenstab of Washington University in St. Louis updated the AAIC audience on Ambulatory Research in Cognition, aka ARC, his group’s smartphone-based testing model. Hassenstab devised this app to tame the huge variability seen with in-clinic cognitive testing. He also aims to replace the exhausting and anxiety-producing in-person testing sessions in the clinic with frequent, short bursts of testing embedded in participants’ daily routine. To do that, Hassenstab has developed an app that allows brief bouts of testing multiple times a day for a week, on a participant’s own phone. “Our strategy is to test often and everywhere, to keep it short, and to combine results of many sessions,” Hassenstab said.
The ARC testing battery includes three tasks that assess working memory, processing speed, and associative memory. Participants complete each one, every day, in four short sessions, and do that for a week. Hassenstab has begun to compare the ARC results to in-clinic tests in Dominantly Inherited Alzheimer’s Network (DIAN) members, and finds that the correlations, while weak, go in the right direction. The ARC tests are highly sensitive to age.
As presented previously, this kind of “burst testing” increases the within-person reproducibility between test sessions (Dec 2017 conference news). In-clinic testing yields retest reliabilities ranging from 0.5 to 0.7, which would be unacceptably low for any other biomarker, Hassenstab noted. His goal was a figure of 0.9 or higher. As of last year, the scientists had not quite reached this goal. But at AAIC, Hassenstab presented a few tweaks to the app, and now, after seven days and 20–22 tests, the tests make the mark on all three tasks. “We can now do as well or better than anything in the clinic,” Hassenstab said.
How do ARC scores relate to brain amyloid and tau deposition? Hassenstab showed new data on a small sample of 39 cognitively normal adults, where he found neither the ARC composite score nor in-clinic testing scores correlated with CSF Aβ42. ARC scores, but not in-clinic tests, did correlate with CSF total tau.
As ARC is being refined, a key aspect continues to pose problems, Hassenstab said. Ideally, participants should take the tests on their own cell phones, but standardizing them on a plethora of devices and operating systems is a challenge. To do that, Hassenstab’s group has developed robots to determine a key variable between devices, which is the time it takes for a screen tap to be recorded in the processor. After testing hundreds of different phones and operating systems, they have learned which phones are suitable and which are not. “Now we can say, you can use this phone, you can’t use that phone,” Hassenstab said.
Tap Dance.
A specially designed robot tests and calibrates touch screen performance on smartphones used in mobile cognitive testing. [Courtesy of Jason Hassenstab.]
Robots aside, a more important question may be how ARC and ORCA compare. ORCA features a large number of items, while ARC has fewer items but administers them rapidly at higher frequency. Baker said, “The two complement each other, and we think we can get some cool data going if we combine efforts.” Hassenstab agreed, saying the groups are already collaborating. “These tests are measuring different things—one is comparing individual learning curves, the other is testing cognition in different environments and situations,” he said.
Yet another group, represented by Kate Papp from Brigham and Women’s Hospital, Boston, presented new data on the clock-drawing test. This time-honored, if dated, cognitive assessment sparked renewed interest when it migrated from paper to digital capture (Dec 2012 news; Dec 2017 conference news). In the traditional form, patients either free-draw or copy the face of an analog clock, with the hands set at 10 minutes past 11, and clinicians score the drawing. In the digital version Papp used, patients draw the clock with a recording pen, which marks its own position every 1.2 milliseconds, generating a continuous steam of data on drawing speed, pauses, location and so on. The data gets compared to a proprietary database of thousands of other drawings by people in different diagnostic groups, and converted to a composite score, ranging from 0 to 100.
The pen’s maker claims that the test, which takes less than five minutes, can tell cognitively normal people from those with MCI or AD. Nowadays, these three categories are no longer nuanced enough, so Papp decided to explore whether the digital pen detects more subtle impairment, and how scores relate to preclinical biomarkers.
Telling More Than Time.
Clock drawing reveals deterioration of cognitive function in people with mild cognitive impairment and dementia. [Courtesy of Kate Papp.]
Her study included 98 people from the Harvard Aging Brain Study who were cognitively normal and had PET scans for amyloid (PiB) and tau (flortaucipir). Their digital clock scores were unrelated to sex or education, but did decline with age. Digital clock performance correlated with performance in other tests of multiple cognitive domains, including processing speed, executive function and memory scores. This suggests the test taps into multiple aspects of cognitive function, Papp said.
The digital clock also picked out subtle cognitive decrements, differentiating people with normal from those with impaired performance on the Preclinical Alzheimer Cognitive Composite (PACC), a measure of mild cognitive decline that is used as an outcome measure in the ongoing A4 secondary prevention trial of solanezumab.
Importantly, scores on the digital clock tracked with biomarkers of amyloid and tau deposition. PiB positivity was accompanied by lower clock-drawing scores, where the amyloid-negative people scored 72/100 and the amyloid-positive people 60/100, for an 18 percent reduction in mean score and an effect size of 1.48. Papp also detected a correlation with test scores when she treated brain amyloid as a continuous variable. Increasingly, researchers are searching for ways to measure the consequences of biomarkers as gradually changing factors in Alzheimer’s disease, not only by comparing groups with artificial demarcations. In a sensitivity and specificity analysis, the five-minute digital clock was as good as the 30-minute PACC at picking out amyloid-positive people, Papp said.
How about tau? Clock drawing was linked to regional tau PET signals in sites of early neurofibrillary tangle deposition in this clinically unimpaired group. Worse scores correlated with more tau in the inferior temporal lobe and entorhinal cortex. Moreover, participants who had both elevated amyloid and inferior temporal tau tended to have even worse digital-clock scores.
Time and Space. Tau deposition selectively affects clock-drawing features. Higher tau correlates with lower scores for proper vertical positioning of a copied clock (top), but not horizontal alignment (bottom).[Courtesy of Kate Papp.]
Going forward, Papp wants to dig more deeply into the wealth of data generated by the digital pen, and the more than 700 features analyzed by the software. For example, in this group, incorrect vertical alignment of a copied clock went hand in hand with higher entorhinal tau, but poor horizontal alignment did not. Does this kind of finding hint something about the underlying biology? Papp also found that the command or copy formats of the test are differentially related to tau deposition. That’s worth further study, she believes. “If these scores are related to biomarkers, we expect we are capturing meaningful information,” she said.
>The next step is to use machine learning to teach the data how to pick out amyloid or tau positivity in larger groups of people. This would open up the prospect of digital clock drawing as a screening tool in the general aging population. It may also be possible to use the digital clock as an outcome measure in prevention or treatment trials. Like the ORCA and ARC developers, Papp, too, needs much more data on how the test performs over time, she said. —Pat McCaffrey
More women have Alzheimer’s than men—but why? At the Alzheimer’s Association International Conference, held July 22–26 in Chicago, speakers agreed that it is not simply because women live longer, but indeed reflects a biological vulnerability. Recent epidemiological data found that women succumb to the disease at younger ages than men do, with higher risk in their late 50s and 60s, but no difference in lifetime AD risk. Scientists reported that women carry a higher tangle burden than do men with the same amyloid load, suggesting that one potential reason for their early onset might lie in a greater vulnerability to tau pathology. This association was primarily driven by APOE4 carriers. Other researchers blamed changes at menopause, when the brain’s glucose use drops. One provocative talk claimed that female brains begin to cannibalize their own myelin for energy during this time. APOE4 carriers appear most vulnerable to this disruption, perhaps leaving their brains marked for future degeneration.
If that sounds disturbing, women can take solace in a bit of good news: New studies of hormone replacement therapy found that newly postmenopausal women indeed can safely take several formulations without harming their cognition. This may reassure women who have been wondering what to do after a much-ballyhooed report in 2003 about ERT and dementia risk in older women scared doctors, women—and funders, for that matter—away from this treatment for some years (May 2003 news). For more on these findings, and other epidemiological data on women, estrogen, and AD risk, read Part 2 of this story.
Overall, scientists said, new data are painting a more nuanced picture of women’s AD risk by tying it to other contributing factors. “Evidence is building that women are at increased risk for neurodegeneration in the face of risk factors such as APOE4 and high amyloid burden,” said Rachel Buckley of Massachusetts General Hospital, Boston. In other words, the vulnerability lies downstream of amyloid accumulation, she noted.
Sex Difference Opens Up At Tau Stage. As brain amyloid load increases, regional tau tangle deposition rises faster in women (red) than in men (blue). [Courtesy of Rachel Buckley.]
Two-thirds of AD patients are women, leading to the commonly held assumption that women run a higher risk of dementia. In Chicago, Arthur Toga of the University of Southern California, Los Angeles, challenged that view with data gleaned from a meta-analysis of 58,000 participants in 27 AD studies. Probing data on the Alzheimer Association’s GAAIN data-sharing platform, Toga and colleagues found no difference in AD risk between men and women overall. That said, women did run a higher risk of developing mild cognitive impairment between the ages of 55 and 70, and AD between 65 and 75. In particular, female E4 carriers were at higher risk than male carriers in this age range (Sep 2017 news).
Other talks drilled down to what might be happening in female APOE4 brains. Buckley noted that cognition declines faster in women than in men who have the same amyloid burden (Buckley et al., 2018). Since tau correlates more closely with cognitive loss than amyloid does, she wondered if tau pathology might explain this. Buckley and colleagues analyzed amyloid and tau PET scans from ADNI and the Harvard Aging Brain Study. The ADNI data comprised 103 cognitively normal controls and 58 people with MCI, while the HABS data set contained 193 cognitively normal older volunteers.
In amyloid-negative cognitively normal people, the researchers saw no sex difference in tau burden. In amyloid-positive people, however, women had more tau signal in their entorhinal cortices than men did, and that gender gap grew wider at higher amyloid burdens (see image above). Similarly, women with MCI had more tau tangles in the inferior temporal lobe than men with MCI did. Notably, the presence of an APOE4 allele boosted a woman’s tau burden more than a man’s, in keeping with Toga’s epidemiological analysis showing higher risk in female E4s. Buckley noted that a larger cohort might well reveal a more dramatic interaction between sex and APOE4.
CSF data reinforced these imaging findings. Previous cross-sectional studies have found that female APOE4 carriers have higher total and phosphorylated tau in cerebrospinal fluid than do men with similar amyloid burdens (Altmann et al., 2014; May 2018 news). Bernard Hanseeuw, also of MGH, wanted to know if tau pathology rises faster in female APOE4 carriers. He analyzed longitudinal CSF samples from an ADNI data set of 239 cognitively normal participants who had provided an average of one sample per year for three years.
In Chicago, Hanseeuw reported that across the whole cohort, total tau (t-tau) and phospho-tau 181 (p-tau) accumulated at the same rate in men and women, as well as in E4 carriers and noncarriers. However, in participants who had brain amyloid, as judged by low CSF Aβ42, p-tau rose faster in female APOE4 carriers than it did in noncarriers and male E4 carriers. This shows that sex, genotype, and pathology interact in ways worth elucidating mechanistically, Hanseeuw said.
Buckley, who collaborated with Hanseeuw, noted that this is the first data showing that tau changes faster in some cognitively normal women than men. She hopes to replicate the finding with tau PET once more longitudinal scans become available. However, Buckley and Hanseeuw both pointed out another possible explanation for more neurofibrillary tangles in women. It could be that men with that amount of tau burden are more cognitively impaired, and are thus not present in studies enrolling cognitively normal people. Men have a higher risk of cardiovascular disease, and vascular cognitive impairment could result in faster cognitive decline, Hanseeuw suggested. Further research should examine this possibility, he added.
Hanseeuw concluded that the current body of data from biomarker and epidemiological studies suggest that women who are at risk for AD, based on APOE4 genotype and amyloid accumulation, accumulate tau tangles faster than at-risk men do. In the absence of amyloid plaques, sex and genotype make no difference. Why tau pathology takes off in at-risk women remains unclear, however. “Further work in the field is important to take into account potential hormonal effects,” Hanseeuw wrote to Alzforum.
Roberta Brinton, now at the University of Arizona, Tucson, reported on one such hormonal study in Chicago. She started out by reminding the audience that the female brain undergoes a massive transition during perimenopause, when estrogen levels plummet. This hormone exerts wide-ranging effects on the brain’s metabolism, including helping it soak up glucose from blood (Apr 2015 news; Nov 2011 news). Studying aging female mice and rats, Brinton found that as their hormone levels dropped, their brains turned to ketone bodies as an alternate fuel source. Astrocytes make ketone bodies from fatty acids. To obtain enough fatty acids, the menopausal brain begins ransacking its own myelin, converting it first to lipid droplets, then to ceramides and fatty acids, Brinton said. In the brains of perimenopausal mice, she sees evidence of disorganized, disrupted myelin sheaths, along with an increase in lipid droplets (Klosinski et al., 2015; Yin et al., 2015).
Does such scary plundering happen in people? Brinton believes it does. In Chicago, she reported seeing evidence for this in a study of 43 cognitively normal women between the ages of 40 and 60, plus 18 men as controls. Women who were going through perimenopause, or were postmenopausal, had about a quarter less brain glucose metabolism, as measured by FDG PET, than premenopausal women did. They also had less active mitochondria, as measured in platelets from blood.
But the biggest change occurred in white matter, as measured by MRI. There, postmenopausal women were a whopping standard deviation below the average value in this cohort. Notably, the drops in metabolism and in white matter occurred in the regions that are affected first in AD, such as the temporal cortex and precuneus (Mosconi et al., 2017; Mosconi et al., 2017).
Brinton noted that this sample was too small to quantitatively parse out APOE4 effects. Even so, just looking at the scans, APOE4 carriers appeared to have the greatest loss of white matter. Menopausal APOE4 carriers also had more amyloid plaques than noncarriers and premenopausal women. Overall, the changes in E4 carriers mimicked those seen in the earliest stages of AD, suggesting their brains might be vulnerable to degeneration. “This could be the initiation of a prodromal phase,” Brinton said.
In ongoing research, Brinton is further examining the E4 effect on white matter in a larger cohort of perimenopausal women she follows. “We’re focused on the impact of the APOE4 genotype on the endocrine aging transition,” Brinton told Alzforum. Besides brain changes, she is also measuring the concentration of ketone bodies in blood samples from this cohort, to try to determine if human brains switch to using this form of energy at menopause as rodent brains do.
Buckley found these data on menopausal changes intriguing and thinks they could explain the differences in tau pathology she sees between older men and women. In cell culture, for example, estrogen levels have been found to modulate the kinases that induce tau hyperphosphorylation. Buckley also speculated that because APOE affects lipid metabolism, the E4 variant may lead to problems handling a potential switch to ketone body metabolism in the postmenopausal brain. She plans to test this hypothesis.
Other evidence further supports the idea that APOE4 carriers are particularly susceptible to deterioration during perimenopause. Brinton noted that in the Early versus Late Intervention Trial with Estradiol (ELITE), postmenopausal women whose metabolic health was worse also performed worse on tests of global cognition, executive function, and memory than did those whose metabolic markers were optimal. For verbal memory, the difference was significant across the cohort, though for the other cognitive tests the difference was significant only in APOE4 carriers. The metabolic markers included measures of blood glucose, cholesterol, triglycerides, ketones, and blood pressure. All of the women were within a predefined normal range on these markers, but those who performed poorly on cognitive tests were on the less-healthy end (Rettberg et al., 2016; Karim et al., 2018). The findings imply that APOE4 carriers with borderline unhealthy metabolism are at the greatest risk for AD-like changes during perimenopause, Brinton said.
Intriguingly, other new research is linking metabolism to white matter. In the August 21 JAMA Neurology, researchers in the U.K. reported that optimal metabolic and cardiovascular health associates with fewer white-matter hyperintensities and better cerebral blood flow in young adults (Williamson et al., 2018). Meanwhile, French researchers report in the same issue that having optimal metabolic markers lowered the risk of developing dementia in people older than 65 (Samieri et al., 2018). These kinds of findings are hopeful, Brinton noted, saying “You can change your metabolic phenotype.” In her perimenopausal cohort, she is investigating whether women with better metabolic health maintain thicker white matter.
Would estrogen therapy help maintain cognition? In a small trial, Brinton and Lon Schneider at the University of Southern California, Los Angeles, gave plant-based estrogen analogs called phytoSERMs or placebo for three months to a cohort of 70 postmenopausal women (Dec 2014 conference news). In this trial, women on drug had fewer hot flashes, and performed slightly better on a complex cognitive task than women on placebo, Brinton said. APOE4 carriers had the largest response to this intervention, she noted. Hot flashes have been linked to white-matter damage (Thurston et al., 2016). Brinton warned against using commercial phytoSERM preparations, however; many are made with soy extract that contains estrogen receptor antagonists, and can actually harm cognition.
Why does estrogen provide so little cognitive benefit to postmenopausal women? Brinton noted that menopause marks the end of the process of disconnecting the brain’s estrogen regulation system. Once that transition has occurred, estrogen will have little effect on brain processes. The best time for hormone replacement therapy might be during perimenopause, Brinton suggested. She is currently testing that hypothesis in perimenopausal rats. Brinton also believes that hormone therapy might be most beneficial in women who are at heightened risk for AD, namely APOE4 carriers with unhealthy metabolism.
In toto, the data highlight a growing realization that not all cases of AD have the exact same etiology, Brinton said. Factors like sex, genetics, and lifestyle can influence how AD develops during aging, and may require different interventions. “With data analytics, we can now parse out different populations of AD patients. We are at the tipping point for a precision medicine, systems biology approach to AD,” Brinton said.—Madolyn Bowman Rogers
The story of estrogen and Alzheimer’s disease is one of controversy. Epidemiological studies consistently suggest the hormone protects the brain, but the landmark Women’s Health Initiative Memory Study (WHIMS) found that hormone replacement therapy harmed cognition in older women who were long past menopause. The data led to the hypothesis that the effect of estrogen depends on age, with younger women benefiting the most. At the Alzheimer’s Association International Conference, held July 22–26 in Chicago, speakers presented the latest data in support of this idea, reporting that hormone replacement therapy given soon after menopause neither helps nor harms the brain. Meanwhile, new epidemiological data presented in Chicago reinforced the notion that a higher lifetime exposure to estrogen preserves cognition, with early menopause significantly boosting dementia risk. These studies still leave unclear how best to protect a woman’s brain from decline. One hint came from a small study that correlated first trimesters of pregnancy with a lower risk of dementia later in life. Because first trimesters exert lasting immunosuppressive effects on women’s bodies, the data suggest that immune regulation could help protect the female brain.
Fifteen years ago, the massive WHIMS study shocked researchers, who had been riding a wave of estrogen’s supposed health benefits, with its finding that oral conjugated equine estrogen (CEE) doubled the risk of dementia, contrary to prevailing data from smaller studies (May 2003 news). However, the women in that study were 65 or older. Researchers were quick to point out that observational data showing a benefit to cognition came from younger women, near the age of menopause (Mar 2006 webinar; Nov 2011 news).
Since then, follow-up studies have borne out this timing hypothesis. Younger women enrolled in the Women’s Health Initiative, who had started hormone therapy between the ages of 50 and 54, had no cognitive deficit compared to those on placebo (Jun 2013 news). Among the older WHIMS participants, women with diabetes suffered the greatest loss of brain volume, suggesting that metabolic health affects whatever estrogen does to the aging brain (Sep 2015 news; Espeland et al., 2015).
In Chicago, Carey Gleason of the University of Wisconsin, Madison, reported the latest data from subsequent trials of hormone replacement therapy. She co-leads the Kronos Early Estrogen Prevention Study (KEEPS), which compared three formulations of hormone replacement therapy administered within three years of menopause to a cohort of 662 women. The therapies were oral CEE, an estradiol patch, or cyclic progestin. After four years, none of these therapies changed any measure of cognition relative to placebo, Gleason reported. However, oral CEE did improve mood, lowering the incidence of anxiety and depression (Gleason et al., 2015). Menopausal women have an increased risk of depression (Bromberger et al., 2011).
Similar findings came out of the Early versus Late Intervention Trial with Estradiol, Gleason reported. ELITE compared women who started oral estradiol within six years of menopause to women who started more than 10 years after. After five years on drug, neither group had any cognitive deficits compared to placebo. Those who started treatment early, however, did have less atherosclerosis than the placebo group, suggesting a benefit to heart health (Hodis et al., 2016).
Gleason emphasized that all the participants in these two studies were in good health, with normal metabolic markers and no chronic conditions. For these relatively young, healthy women, short-term hormone therapy has no effect on cognition, Gleason concluded. At the same time, she hinted that estrogen could have distinct effects on subgroups of women. In a small substudy, Gleason found the amyloid burden on PiB PET to be lower in 21 KEEPS participants who used the estradiol patch than in 30 women on placebo. The difference seemed to be driven by ApoE4; the 10 carriers using the patch accumulated less amyloid than the five on placebo (Kantarci et al., 2016). An ongoing KEEPS continuation study will follow up with participants 12 years after they started therapy to check for long-term effects, and will examine amyloid burden in a larger sample.
While estrogen’s benefit for old brains is in doubt, the hormone’s importance for younger women seems unquestioned. In Chicago, Paola Gilsanz of the University of California, San Francisco, strengthened the case that lifetime exposure to estrogen lowers AD risk. She analyzed data from 14,595 women between the ages of 40 and 55 who were seen in Kaiser Permanente clinics in California between 1964 and 1973. The cohort was diverse, with 68 percent white, 16 percent African-American, 6 percent Asian, and 5 percent Latina in the mix. The women had an average of three children and one miscarriage each, and an average reproductive period of 34 years. The average age of menarche was 13, and menopause, 47. The researchers assessed for dementia by examining medical diagnostic codes up through 2017, a follow-up of about 50 years. They adjusted all outcomes for age, race, education, and various health factors known to affect a woman’s AD risk.
Overall, 36 percent of the cohort developed dementia, but a woman’s risk dropped the more children she had. Compared to women with one child, those with three or more had a 12 percent lower risk. Estrogen levels surge during pregnancy, Gilsanz noted. On the other hand, having even one miscarriage boosted the odds of dementia by 25 percent. A late onset of puberty or early onset of menopause, leading to a short reproductive period, increased risk even more, by 31 and 28 percent, respectively. In general, every additional year in a woman’s reproductive span lowered her odds of AD by 2 percent, Gilsanz said.
The findings suggest that the more estrogen a woman is exposed to during her lifetime, the better her brain fares, Gilsanz said. On the other hand, miscarrying may indicate an unfavorable hormonal milieu, or other underlying health problems, she suggested.
The evidence suggests that estrogen strongly protects women’s brains before the age of 50, but does so only moderately between the ages of 50 and 59, and perhaps becomes harmful at age 60 and after, Walter Rocca of the Mayo Clinic in Rochester, Minnesota said in Chicago.
Another small study of women’s reproductive health drew a different conclusion. Molly Fox, University of California, Los Angeles, analyzed a case-control cohort of 133 women between the ages of 70 and 100 in England, half of whom had been clinically diagnosed with dementia. Fox looked for any correlation between reproductive history and dementia, and found that the more months a woman had been pregnant in her lifetime, the less likely she was to have dementia. Each additional month of pregnancy reduced risk by 5 percent, Fox reported in Chicago. If this was due to estrogen exposure, lower risk should correlate with the number of third trimesters, when estrogen spikes, Fox reasoned, but she found no such relationship. Instead, the number of first trimesters appeared protective, with each additional one lowering AD risk by about 30 percent (Fox et al., 2018).
Why might that be? During the first trimester, the body boosts the number of regulatory T cells that suppress immune reactions to lower the odds of rejecting the fetus. If this immunosuppression persisted long-term, Fox suggested, it might limit inflammatory damage late in life. Inflammation contributes to AD and other neurodegenerative diseases. “Pregnancy may reorganize the body in ways that protect against AD later,” Fox said. She acknowledged her tiny study serves mostly as a stimulus to do this kind of analysis in larger cohorts, with examination of immune biomarkers. Intriguingly, a recent study found that microglia in aging female mice change their gene expression more than do microglia in aging males, strengthening the idea that the immune system may contribute to the female vulnerability to dementia (Aug 2018 news).—Madolyn Bowman Rogers
With Sudden Progress, Blood Aβ Rivals PET at Detecting Amyloid
A lot can happen in a year. At the 2017 Alzheimer’s Association International Conference in London, Randall Bateman’s team at Washington University in St. Louis wowed the crowd with a blood assay for Aβ that predicted brain amyloid with previously unattainable specificity and sensitivity (Jul 2017 conference news). Coming after nearly two decades of trying, without much success, the mass spec-based technique reignited the quest for a blood test for Alzheimer’s pathology that would equal the diagnostic gold standards of cerebrospinal fluid testing and amyloid PET scans, but less invasively and cheaper, respectively. Soon after, a Japanese group debuted a second highly accurate mass spec technique (Feb 2018 news), and by AAIC 2018, held July 20–26 in Chicago, a competitive field was on full display.
There was news of a high-sensitivity immunoassay that would be simpler than mass spec and equally good at predicting amyloid positivity. There was a first stab at screening blood with an automated ELISA platform. Scientists presented new data from existing ELISA and mass spec platforms, and an ultrasensitive approach that uses magnets to detect antibody-Aβ complexes on spinning beads. With results rolling in, leaders in the field are already thinking about how to standardize plasma assays, and looking to the pathway they used to bring order to CSF testing (Hansson et al., 2018).
“Blood tests were the biggest story at AAIC this year,” said Michael Weiner of University of California, San Francisco. “They will become widely available and revolutionize the field in the next few years,” predicted Colin Masters of the University of Melbourne, Australia, who was involved in vetting one of the mass spec assays. “The future vision is that you have the blood test periodically, maybe annually once you are in your 50s, and when your Aβ42/40 ratio changes over two years, then you do something. It could be a trial, a BACE inhibitor, or another safe preventive drug.”
Alzheimer’s disease is defined in part by brain amyloid, which shows up in living people on a PET scan, or can be inferred by a drop in CSF Aβ42 or the Aβ42/40 ratio. But PET is expensive and uses radioactivity, and lumbar punctures are invasive. Neither is quite right for screening and frequent repetitive measures in a clinical trial, or for prescreening in the routine course of healthcare. Indeed, recent early stage trials that enrolled based on biomarkers and no, or only subtle, cognitive signs have struggled with unacceptably high screen failure rates of up to 80 percent.
A blood test would be ideal: With a simple draw, people could have a measure of their likelihood of harboring brain amyloid. While that information could be used to enrich clinical trials, the main use of a blood test would be widespread screening in the general population for brain amyloid, said Philip Scheltens, VU University, Amsterdam. “People deemed negative would be sent on their way, while those in the positive range could be candidates for additional testing, possibly including PET and CSF,” he told Alzforum.
The mass spec assays are an option, though samples will have to be sent to central labs for analysis, and some researchers wondered whether they can be scaled up sufficiently for widespread clinical use. Detection by ultrasensitive antibody-based single-molecule array (Simoa) offers an alternative. The Aβ42 and Aβ40 Simoa assays from Quanterix in Lexington, Massachusetts, have proven to reliably pick out the vanishingly low picogram per ml concentrations of the peptides present in plasma. Using this assay, Swedish researchers previously documented lower plasma Aβ40 and 42 in people with AD dementia than controls; people with mild cognitive impairment had intermediate levels. Alas, where CSF Aβ concentrations robustly distinguished amyloid-positive and -negative people in the early, presymptomatic stage of disease, plasma Aβ did not (Janelidze et al., 2016).
In new data presented at AAIC, Inge Verberk at VU University used a Quanterix Simoa test to measure both plasma Aβ peptides and tau in a different group of 248 elders with subjective cognitive decline and known amyloid status, taken from the Amsterdam Dementia cohort. Consistent with the Swedish results, amyloid-positive people had lower plasma Aβ42 and Aβ42/40 ratios compared with amyloid-negative participants. In Verberk’s study, however, the ratio was able to distinguish amyloid status with moderate sensitivity and specificity, with an area under the curve (AUC) of 0.76. Importantly, lower Aβ42/40 was associated with a nearly twofold greater risk of clinical progression to MCI or AD during follow-up. The results suggest that blood assays have prognostic value early in AD, though there is room for improvement.
Better, Best? In a side-by-side comparison of three plasma Aβ assays, a new prototype on the Simoa platform reportedly achieved higher accuracy than the commercially available Simoa (Quanterix) or the Euroimmun ELISA. [Courtesy of Teunissen lab.]
Elisabeth Thijssen, who works with Charlotte Teunissen at VU University, presented a promising new version of the Simoa for Aβ40 and 42. Working in collaboration with ADx Neurosciences, Ghent, the scientists switched up the antibodies that are used to capture and detect Aβ42 and 40, and fiddled with the assay conditions to optimize the test for plasma. To pull peptides from serum, they chose the monoclonal antibodies 21F12 and 2G3, which bind to C-terminal residues of Aβ42 and 40, respectively. For detection of the immobilized peptides, they used the pan-peptide 3D6 antibody, which binds to the first five N-terminal residues of both Aβ42 and 40. This differs from the commercially available Quanterix assay, which uses an N-terminal antibody to capture both peptides, followed by 21F12 or 2G3 to detect Aβ42 and 40, respectively.
Thijssen compared the ratio of plasma Aβ42/40 in samples from 20 amyloid-positive AD patients and 20 amyloid-negative people with subjective memory complaints who were matched for sex and age. Amyloid status was assigned on the basis of CSF testing. Because the readings were more tightly clustered in each group, this new assay produced larger group differences and less overlap between groups than a standard ELISA using the same antibodies, or indeed than the Quanterix Simoa kit. The tighter numbers gave a better diagnostic performance: Thijssen showed 95 percent sensitivity and specificity at distinguishing controls from AD for her prototype, compared with 85 and 86 percent for the Quanterix Simoa kit or ELISA assays, respectively. Of the three, the prototype assay also produced the best correlation between Aβ42/40 in plasma and CSF.
In this first feasibility study, the optimized assay performed better than the Quanterix kit at distinguishing amyloid-positive from -negative, with an AUC of 0.953 vs 0.852 for the kit, Thijssen reported. The performance was also slightly better than the mass spec assay from the Washington University group (Ovod et al., 2017), and comparable to the top-performing test so far, the Shimadzu mass spec procedure.
Going forward, the Amsterdam group will conduct a larger study on about 400 people, including healthy controls and people with MCI and AD who have amyloid PET data available. Those results will indicate whether the plasma assay correlates with PET, and if it can be used as a screening tool, Thijssen and Teunissen said.
In parallel, ADx NeuroSciences will continue to develop and validate the assay format, including scale-up. Both groups will collaborate to commercialize the assay, which generated intense interest at the meeting from researchers involved in many of the large clinical cohorts, Teunissen told Alzforum. The talk also raised the interest of representatives from the Bill Gates Foundation’s new Alzheimer’s initiative (Jul 2018 news), who were spotted chatting with Thijssen and Teunissen after the session.
Existing plasma assays are getting new attention, as well. Araclon Biotech of Zaragoza, Spain, offered a blood test for Aβ peptides four years ago, but at first found few takers, Araclon’s Ian Sherriff told Alzforum. Their immunoassay tots up free Aβ42 and 40, as well as what they call “total Aβ,” a fraction that includes peptides bound to plasma proteins and red blood cells. The bound fractions are not detected in ELISA or Simoa assays. Araclon scientists originally developed the assay to help along the clinical trial of their anti-amyloid antibody, and then began to offer analysis on a contract service basis. Sherriff told Alzforum that this business has been picking up in the past year.
“It was difficult to convince people we had a decent signal,” Sherriff said. “Now that other groups are coming out with data, that helps a lot. The whole mindset in industry is that blood-based biomarkers are back in,” he told Alzforum.
At AAIC, Shannon Risacher from Liana Apostolova’s lab at Indiana University School of Medicine, Indianapolis, showed how she used the Araclon service to compare blood Aβ to imaging measures of amyloid, tau, and neurodegeneration. She compared Aβ42 and 40 in 47 volunteers who had structural MRI and amyloid PET imaging; 41 also had tau PET with flortaucipir. The group included 20 cognitively normal people, 11 with MCI and 16 with AD. As expected, Aβ42/20 ratios were reduced in patients with AD. The Araclon test distinguished amyloid-positive and-negative in the full sample, with an AUC of 0.8. Among cognitively normal people, the separation was better, with an AUC of 0.865.
Looking at regional amyloid accumulation, Risacher found that a lower plasma Aβ42/40 ratio was linked to increased brain amyloid across the cortex, most strongly in the frontal, parietal, and temporal lobes. The study showed, for the first time, an association of blood Aβ with tau: In this group, total Aβ42/40 negatively correlated with total levels of cortical tau, and more specifically with tau deposition in lateral temporal, inferior parietal, and parietal regions. The ratio’s association with neurodegeneration was much weaker. Risacher detected trends, but no significant association with hippocampal CA1 or CA2-3 volume or subiculum volume. “This shows an association of blood Aβ with specific pathology,” she told Alzforum. “It’s strongest with amyloid, next with tau and weakest with neurodegeneration, which again suggests more sensitivity early in the disease process,” she said.
Risacher would like to repeat her analysis in a larger sample, but also thinks this initial group offers an opportunity to compare different assays head to head. She has sent samples to Henrik Zetterberg, University of Gothenburg, Sweden, for analysis with his lab’s Simoa assay.
Researchers in Koichi Tanaka’s mass spectrometry research lab at Shimadzu Corporation, Kyoto, Japan, continue to refine their method, which combines immunoprecipitation of Aβ peptides with quantitative MALDI-TOF mass spec. As published earlier this year, a composite biomarker based on Aβ42 and 40 blood concentration was 87.5 percent concordant with PiB PET, using a mean cortical SUVR of 1.4 as the cutoff for positivity. At AAIC, Akinori Nakamura in the Tanaka lab presented new data on discordant cases, where PET and blood measures disagreed. He focused on 10 cases where the SUVR was above 1.30 but just shy of the cutoff. All 10 were deemed positive by visual read of their PET scan, and six were classified positive based on the blood biomarkers. Seven of the 10 had follow-up imaging within 12 to 60 months, and by that time four of them crossed the SUVR cutoff. These results indicate that the plasma biomarker is sensitive enough to detect subthreshold amyloid accumulation, Nakamura concluded.
That jibes with data presented by Sebastian Palmqvist, Lund University, Sweden. Palmqvist ran a cross-sectional analysis of changes in a host of biomarkers in the Swedish BioFINDER study, showing that CSF Aβ42 dropped first, followed by the Aβ42/40 ratio. Decreases in plasma Aβ42 and the 42/40 ratio trailed closely behind, at a time when PET signals were ramping up in early amyloid-accumulating regions, but had not yet crossed the threshold of brain-wide amyloid positivity.
Naoki Kaneko of Shimadzu said the Tanaka lab is planning collaborations with sites in the U.S. and Europe to further test their assay. The company is planning to offer commercial analysis services, possibly within this year, he said.
Another entrant into the plasma biomarker arena, MagQu, New Taipei City, Taiwan, markets a unique platform to assay plasma Aβ42, 40, tau, p-tau and α-synuclein. Their system uses a single antibody conjugated to magnetic nanobeads to capture each protein, and a magnetic spin technology to separate and detect antigen-antibody complexes (Apr 2018 conference news). Other available ultrasensitive assays use two antibodies, or one antibody and mass spec for detection. Unlike the sandwich assays or mass spec that detect a decrease in plasma Aβ42 in people with AD dementia, the MagQu assay finds plasma Aβ42 increased or unchanged (Teunissen et al., 2018; Lue et al., 2017). “This is puzzling, but appears very reproducible,” Zetterberg told Alzforum. He speculated that the MagQu assay may be detecting Aβ aggregates and/or Aβ bound to other proteins. Consistent with this idea, Korean researchers recently reported an increase in plasma Aβ42 in people with AD using an oligomer-specific ELISA (Wang et al., 2017). Teunissen told Alzforum her group is planning a side-by-side comparison of that ELISA with the MaqQu and her new Simoa. “Hopefully, that will provide a more conclusive comparison of value of the tests,” she said.
At AAIC, Lih-Fen Lue of Arizona State University showed new data using the method to analyze plasma Aβ42, 40, and tau in 368 healthy people from ages 24–91 from Taiwan, China, Sweden, and the U.S. Plasma Aβ42 and 40 showed a slight decrease with age, but the correlation was weak. She did see a correlation between Aβ42, 40, and t-tau that appeared in middle age and was strongest at ages 60–69. The company has opened a sample testing lab in Arizona to provide the assay in the U.S.
The Global Biomarkers Standardization Consortium (GBSC), led by University of Gothenburg’s Kaj Blennow and convened by Alzheimer’s Association, has led the effort to standardize CSF biomarkers, and in Chicago, the group considered moving quickly to do the same for plasma biomarkers. Michelle Mielke, Mayo Clinic, Rochester, Minnesota, leads the Association’s Biofluid-Based Biomarkers interest group; she updated the GBSC on plasma biomarkers at their preconference, where there was also a panel discussion on plasma Aβ. “There is a lot of interest in plasma Aβ. We need to figure out who is working on what, so we can identify means of collaborating and not recreating the wheel. To my knowledge, there is no consensus yet on what platform should be used for plasma amyloid, but if we want to use it as a screening measure, it does need to be made to be high throughput,” Mielke told Alzforum.
Could one such assay be the Elecsys fully automated immunoassay? Oskar Hansson at Lund University had previously shown that Roche’s automated assay for CSF Aβ42, total-tau, and phospho-tau181 was just as accurate at assigning amyloid status as a visual read of an amyloid PET image by an experienced clinician (Hansson et al, 2018; Apr 2017 conference news).
To find out how the automated immunoassay would work with blood, Hansson collaborated with Roche to put the system through its paces on 843 plasma samples from the Swedish BioFINDER study. Roche slightly tweaked the assay to optimize it for the lower measuring range of plasma, and incorporated plasma-specific calibrators and controls. Hansson then analyzed biomarkers from CSF and plasma side by side for 513 cognitively unimpaired participants, 265 with MCI, and 65 with AD dementia. Plasma Aβ42/40 ratios were lower in amyloid-positive than amyloid-negative people, regardless of their cognitive state, and the combination of plasma Aβ40 and 42 predicted brain amyloid status with an AUC of 0.80. The accuracies were similar when scientists looked separately at cognitively healthy elderly, or at patients with subjective cognitive decline or MCI.
A composite biomarker comprising plasma Aβ42, Aβ40, and ApoE genotype did a little better, achieving an AUC of 0.85 for brain amyloid status. That was lower than CSF, but higher than the guess of specialists based on a clinical exam and MRI structural imaging of people with subjective cognitive decline (SCD) or MCI (AUC=0.65). Some simple cognitive tests add accuracy to this composite, Hansson said. “Overall, the current sensitivity and specificity of Elecsys plasma tests is not good enough to replace CSF/PET biomarkers, but it was better than the diagnosis by specialist clinicians assessing their patients with SCD or MCI,” Hansson said.
Alas, in primary care, diagnostic accuracy of AD is even lower, Hansson said. He hopes to optimize the Elecsys plasma assay for use in primary care. Even at its current level of sensitivity and specificity, he thinks it could be useful to help primary care practitioners decide whom to refer for specialist neuropsych assessment and CSF or PET testing. However, this needs to be further evaluated in large numbers of people recruited in primary care centers who are less likely to be amyloid-positive, he said.
Tobias Bittner of Roche noted an urgent need for standardization of sample handling for the plasma tests. He said Roche is working on that now, gathering data on the stability of Aβ peptides in plasma and other parameters. “We all need to understand how Aβ behaves in plastic, whether you can transfer from tube to tube or freeze and thaw, the optimal time to centrifuge blood, etc. Preanalytical standardization was a huge problem for CSF, and now we need to look at that for plasma,” he told Alzforum. Bittner said that much of the work is done and promised to share results soon.
Zetterberg and Blennow are coordinating a round-robin study involving all available plasma Aβ methods, in which aliquots of the same samples will be sent around to labs performing the different assays. “We can then compare how well they correlate and what their variations are. We will also examine their potential diagnostic utility by calculating the ratio of amyloid-positive and -negative samples, with amyloid positivity determined by PET,” Zetterberg wrote in an email to Alzforum. They aim to include samples from 20 amyloid-positive and 20 amyloid-negative people. The challenge is to get large enough volumes to provide aliquots for all the labs, he said.—Pat McCaffrey
Brain Damage from Cardiovascular Disease Starts Earlier Than You Think
Scientists agree that midlife cardiovascular risk factors portend late-life dementia. But exactly when in midlife? Two separate, but complementary, studies presented at this year’s Alzheimer’s Association International Conference, held July 22–26 in Chicago, come down firmly in the 40s and even earlier.
Matthew Pase, Swinburne University of Technology, Hawthorn, Australia, collaborated with Charles DeCarli at University of California, Davis, to analyze data from the Framingham Heart Study. Pase reported that no matter how long a person lived, his or her risk for future brain atrophy due to cardiovascular disease had always been highest when they were about 45. “The study suggests that the effects of cardiovascular risk factors on the brain are much more robust in the 40s and then diminish as people get older,” said DeCarli.
Similarly, Christopher Lane, from University College London, England, reported that in a cohort followed from birth, high blood pressure at age 53, and rising blood pressure in the decade before that, correlated with damage to small blood vessels in the brain at about age 70. Hypertension in the 40s also correlated with later-life brain atrophy. In marked contrast, Lane found no link between blood pressure and accumulation of brain amyloid. “The relationship between midlife hypertension and vascular disease was expected. What was less clear was the epoch when that risk was strongest, that acceleration as well as absolute levels of blood pressure was important, and that the effect of blood pressure was on small vessel disease and brain volume but not amyloid accumulation,” noted Jonathan Schott, who led the UCL study.
Vascular risk factors in the 40s and 50s, including hypertension, have been linked to later-life dementia (Aug 2017 news; Jun 2018 news). Others had previously reported that the sooner in life blood pressure rises, the stronger it associates with late-life structural damage in the brain. Most of those studies have been limited to specific populations (Swan et al., 1998; Power et al., 2016; Knopman et al., 2011; Debette et al., 2011).
Schott and colleagues study a birth cohort that is representative of the whole of mainland Britain. It has been followed prospectively since 1946. At that time, the British government was concerned that population growth would be insufficient to support rebuilding the country after the decimation of World War II. They commissioned a survey of every birth in the U.K. during a single week, March 3 to 9, all 13,687 of them. Then, recognizing an opportunity to learn how the environment shapes mental and physical development, the government decided to track a representative sample of those babies, following 5,362 singletons who had been born to married parents. The National Survey of Health and Development has since seen and tested the men and women of this cohort 24 times.
In the early days, the survey took stock of educational attainment, socioeconomic status, diet, exercise, and mental health. In 1982, when the cohort were 36 years old, they began receiving more extensive clinical tests, including blood pressure measurements and tests for respiratory, cognitive, and musculoskeletal function. Between ages 60 and 64, members of the cohort were offered more intensive cardiac screening. About 1,200 accepted and underwent EKG, echocardiograms, and Doppler ultrasound of the carotid arteries to look for arterial plaques. “We know an enormous amount about these volunteers,” said Schott.
Representative Sample. Distribution of volunteers in Insight46 (black dots, left), reflect the population density of the U.K. (right). [Courtesy of Jonathan Schott.]
To learn about their brain health, Schott, together with Nick Fox and Marcus Richards at UCL, began the Insight46 study (Lane et al., 2017). This sub-study selected 500 of the people who had volunteered for more intensive cardio testing in their early 60s. It plans to scan their brains with structural MRI and florbetapir amyloid PET twice, two years apart. Baseline assessments began in 2015 and were finished in January of this year. Insight46 also collects clinical, neuropsychological, fluid biomarker, and genetic information. In his talk at AAIC, Lane focused on how the imaging data relate to blood pressure.
Lane wanted to know how three measures—brain amyloid, brain volume, and white-matter lesions reflecting damage to the brain’s teeny blood vessels—relate both to absolute blood pressure values and their rate of change. Blood pressure had been taken when the cohort was 36, 43, 53, 60–64, and 69 years old. Systolic blood pressure (SBP) steadily rose during this time from about 125mmHg to 134mmHg for men, and from about 115mmHg to 131mmHg for women. Diastolic pressure (DP) rose initially, too, from about 81mmHg to 86mmHg for men and from 76mmHg to 80mmHg for women, but began to fall at age 53 to end up at about 74mmHg for men and 73mmHg for women by age 69. How would these numbers relate to brain health?
Associations were extremely narrow. Lane reported that only at age 53 did higher SBP and DBP correlate with the extent of white-matter hyperintensities (WMHs) at 69, even though the SBP held steady from age 53–69. The change in both SBP and DBP between ages 43 and 53 correlated with WMHs as well, but the change over other decades did not. What does this mean? The idea is that having higher blood pressure during that specific decade puts people at risk for brain damage in later life.
Analysis of whole brain volume told a similar story, albeit the risk might come sooner. Lane reported that higher DBP at age 43, and the change in DBP from age 36–43, correlated with smaller brain volumes.
Lane thinks it important not only that the absolute blood pressure tracked with brain damage, but also the change in pressure. “We know high blood pressure is bad for cardiovascular and cerebrovascular health, and the benefits of screening are well recognized, but that does not take account of previous blood pressure over time,” he told Alzforum. “We should be thinking of monitoring change as well as the absolute number,” he said. This could be done with wearable devices, Lane suggested.
DeCarli said the study complemented many others in the field. “Drawn from an entire population across mainland Britain, the birth cohort confirms and extends what others have shown, namely that the effect of cardiovascular disease on brain health is stronger in midlife,” he told Alzforum.
That said, DeCarli thinks Insight46 underestimated the effect, because the MRI measurements were taken when the volunteers were in their 70s. He, Pase, and colleagues correlated cardiovascular risk with brain lesions both cross-sectionally and longitudinally in participants of the Framingham Heart Study. In 1948 the FHS began to track the cardiovascular health of some 5,000 volunteers, aged 30–59. It is now in its third iteration, having recruited children and grandchildren of the original cohort in 1971 and 2002, respectively. Volunteers began having brain MRI scans around 1999. Pase correlated brain volume measured by MRI with scores on the Framingham Stroke Risk Profile, a validated and widely used measure of overall cardiovascular risk based on a plethora of parameters including, age, gender, cardiovascular disease, atrial fibrillation, diabetes, blood pressure, and tobacco smoking history.
At AAIC, Pase reported that in cross-sectional analysis, higher stroke risk correlated with smaller brain volume at that same point in time. The association was strongest in those aged 45. Similarly, when he looked prospectively at how stroke risk relates to future brain volume, he again found the greatest threat came in the 40s, no matter how old the person became. For example, in those aged 85, brain volume more tightly correlated with stroke risk at age 45 than at age 55, 65, or 75 (see image below). The same was true for 75- and 65-year-olds—in each case the strongest risk for brain atrophy came at age 45. The effect was even stronger in women.
Midlife Crisis. On average, cardiovascular risk for late-life brain volume loss (beta estimates of effect size) is strongest at age 45, no matter when the atrophy occurs. [Courtesy of Matthew Pase.]
Schott thinks the Framingham data dovetails with his group’s discovery in the U.K. cohort. “While they used a composite of vascular risk rather than blood pressure, they showed that, the further in time from the brain volume measure, the stronger the association. They found a very similar effect,” he told Alzforum.
To Lane, this is an eye-opener. “Midlife is often thought to be from 40 onwards, but we see effects on the brain from age 36–43. We need to consider the risk of high blood pressure earlier in life,” he said. Alas, DeCarli thought that finding curious. “We have looked at 30-year-olds in the Framingham study, and find very few who have hypertension. It is much more common by age 50, when 25 percent of the population are affected,” he said. DeCarli added that people who have hypertension already in their 30s are in big trouble. “I suspect many of those people do not even survive to their 70s,” he said.
Alzheimer’s or Dementia?
Regarding amyloid, a link between midlife hypertension and plaques later in life has been harder to pin down. Some postmortem and PET studies have suggested a correlation (Petrovitch et al., 2000; Langbaum et al., 2012), but David Knopman, Mayo Clinic, Rochester, Minnesota, told Alzforum that both the Atherosclerosis Risk in Communities (ARIC) study and the Mayo Clinic Study on Aging have found no link between hypertension and brain amyloidosis. “I am glad that the UCL group came to the same conclusion,” Knopman said. In ARIC and other cohorts, researchers have reported a link between arterial stiffness, often an underlying cause of hypertension, and the accumulation of Aβ in the brain (Hughes et al., 2018; Hughes et al., 2013).
That is not to say high blood pressure poses no risk for Alzheimer’s disease. “That high blood pressure does not influence accumulation of Aβ does not, however, mean it does not influence AD,” stressed Schott. “It could affect other downstream pathologies associated with the disease, such as neuronal loss or inflammation.” In fact, results from the SPRINT MIND trial presented at AAIC show that for 60-year-olds with hypertension, lowering their systolic blood pressure to about 120 mmHg reduced incidence of mild cognitive impairment by 19 percent over 3.26 years (Aug 2018 conference news).
Schott noted that even a country-wide birth cohort study has its limitations. For example, Insight46 thus far has no postmortem or tau PET data, though 60 percent of the cohort have agreed to donate their brains.—Tom Fagan
Lane CA, Parker TD, Cash DM, Macpherson K, Donnachie E, Murray-Smith H, Barnes A, Barker S, Beasley DG, Bras J, Brown D, Burgos N, Byford M, Jorge Cardoso M, Carvalho A, Collins J, De Vita E, Dickson JC, Epie N, Espak M, Henley SM, Hoskote C, Hutel M, Klimova J, Malone IB, Markiewicz P, Melbourne A, Modat M, Schrag A, Shah S, Sharma N, Sudre CH, Thomas DL, Wong A, Zhang H, Hardy J, Zetterberg H, Ourselin S, Crutch SJ, Kuh D, Richards M, Fox NC, Schott JM.
Study protocol: Insight 46 - a neuroscience sub-study of the MRC National Survey of Health and Development.
BMC Neurol. 2017 Apr 18;17(1):75.
PubMed.
When axons in the brain’s white matter start to fall apart—be it from insult or injury—they release the protein neurofilament light chain. Thus dumped, NfL finds its way into the cerebrospinal fluid and into the blood. Rising CSF NfL signals neuron loss in acute and chronic conditions, including stroke, traumatic brain injury, Alzheimer’s, multiple sclerosis, and others. Now, the advent of a supersensitive assay for blood NfL has led to an explosion in studies evaluating the protein as a biomarker for neurodegeneration. New data spilling out at the Alzheimer's Association International Conference, held July 20–26 in Chicago, offer a glimpse at the progress. The data is so convergent that consensus is already forming that blood NfL can help predict a person’s disease course. Besides helping to stage participants in clinical trials, NfL might serve both as a dynamic marker of neuronal injury and as a tool to measure treatment effects in trials.
Scientists already knew that NfL was consistently higher in the CSF of people with AD than controls (see AlzBiomarker meta-analysis). It tracks with prognosis, whereby more elevated concentrations presage faster disease progression (Nov 2015 news).
Now, scientists are realizing that as goes CSF, so goes blood. And that opens new doors. For years, reliably measuring NfL in blood, where concentrations run 50 times lower than CSF, was impossible. Then Henrik Zetterberg, University of Gothenburg, Sweden, developed an ultrasensitive NfL immunoassay on the Quanterix Simoa platform (Apr 2016 conference news; Kuhle et al., 2016). His and other groups’ work (Bacioglu et al., 2016) established that blood NfL concentrations mirror those in CSF. In a cross-sectional study looking at plasma NfL in 570 ADNI participants, blood NfL, just like CSF, appeared to faithfully report disease severity and prognosis (Mar 2017 news).
At AAIC, a large, longitudinal study addressed the next question, that is, how blood NfL changes over time. Stephanie Schultz, Washington University, St. Louis, presented data on serum NfL in more than 400 people in the Dominantly Inherited Alzheimer’s Network. DIAN offers a unique opportunity for biomarker investigation, because mutation carriers are destined to get AD, and researchers know approximately when they will develop symptoms, based on their specific genetic lesions and family history. Mathias Jucker’s group at the German Center for Neurodegenerative Disease in Tubingen, Germany, measured NfL using the Simoa assay, and Schultz and her colleagues did the statistical analysis in the cohort, who have been followed for years with comprehensive phenotyping, including repeated MRI and PET imaging, and CSF and blood sampling. All had given more than one blood sample; half had given more than two, collected every one or two years. The mostly short stretches of within-person longitudinal follow-up added up to span 30 years of Alzheimer’s progression from presymptomatic to advanced dementia.
First, Schultz and colleagues ran a cross-sectional analysis. It substantiated earlier findings from a much smaller ADAD cohort in the U.K. (Nov 2017 news). In both cohorts, mutation carriers had on average higher CSF and blood NfL concentrations than noncarriers, regardless of their cognitive status. This ongoing ADAD research is also measuring serum NfL as a function of time to symptom onset. Schultz and colleagues are finding that the difference emerges five to 10 years before the estimated age of onset (EYO), the same as when NfL was measured in CSF, Jucker wrote to Alzforum.
For most biomarkers being studied, scientists are realizing that longitudinal measures of a given marker’s rate of change are more sensitive than just the baseline concentration at predicting cognitive decline (e.g., Dec 2017 conference news). Schultz and colleagues found the same for NfL. In the longitudinal part of the study, the rate of change of NfL held steady over time in noncarriers, but appeared to quicken in presymptomatic carriers approximately 16 years before onset of symptoms, or about 10 years earlier than estimates from the cross-sectional analysis. The rate of change appeared to peak at the transition from normal to dementia, then plateaued.
When compared with imaging biomarkers, the rate of change of serum NfL was associated most strongly with cortical thinning, and less so with declines in FDG-PET or Aβ accumulation. This suggests the increase in blood NfL reflects ongoing neurodegeneration. Consistent with that, the rate of NfL change also predicted cognitive decline—in a subset of 39 people, speedier NfL accumulation tracked with greater decline in MMSE and logical memory in the following one to two years.
Blood NfL increased fastest in those transitioning from cognitively normal to symptomatic, and this rate was more sensitive to predicting a person’s progression than their absolute NfL levels. These findings highlight the value of longitudinal data, Schultz said. The next logical step, she said, will be to relate NfL to measures of white matter and to synapse integrity.
“Our work suggests that NfL in blood is going to be useful as a strong and clear signal of neurodegeneration,” said co-author Brian Gordon of Washington University. DIAN is adding plasma NfL as a core biomarker, so it will be a standard measure moving forward, he added.
At AAIC, Philip Weston,University College London, showed longitudinal data on his center’s ADAD cohort. It agreed closely with Schultz’s findings. Among the 15 mutation noncarriers and 47 carriers Weston follows, 41 thus far have given blood repeatedly, for a total of 118 time points. As published before, baseline NfL was higher in symptomatic people, 22.6 pg/ml in the AD group versus 12.6 in asymptomatic carriers and 10.34 in noncarriers. More strikingly, though, Weston detected a dramatic escalation of the annual rate of change of NfL from 0.51 and 0.67 pg/ml/year in noncarriers and asymptomatic carriers, respectively, to 3.52 pg/ml/year in the five symptomatic people who had given multiple samples.
When he modeled the longitudinal changes versus time of onset, blood NfL concentration appeared to diverge between carriers and noncarriers around a decade prior to onset of clinical symptoms, and rose progressively after. Raquel Sanchez-Valle, Barcelona University Hospital, presented additional data in her Spanish ADAD cohort that supports the idea that serum NfL is a good proxy for tracking disease onset and progression in this group.
Weston had too little data to say what happened in the years after onset, but he did notice a difference with type of mutation: Presenilin mutation carriers had 34.5 percent higher NfL than APP mutation carriers. Weston emphasized that in this small sample, he sees great inter-individual variability in the rate of change, which may limit NfL’s usefulness in tracking individuals. Other factors can affect NfL; Its concentration creeps up with age, with vascular disease, and with neurodegenerative processes besides AD. “That’s something we need to think about more,” he said.
Like ADAD, like LOAD?
How these results in ADAD will apply to sporadic AD remains unclear. “I don't know of any studies that directly compared NfL in sporadic and familial AD. This is extremely important in knowing the wider applicability of these markers,” Weston said. The ADNI NfL study reported no difference in blood NfL between asymptomatic amyloid-positive and -negative people, but it was not clear how close those people might be to developing symptoms. “Perhaps they were too early in the disease process,” Weston said. Gordon told Alzforum he hopes to measure plasma NfL in WashU’s sporadic AD cohort.
Scientists at the Mayo Clinic in Rochester, Minnesota, are starting to widen the lens by analyzing NfL in the Mayo Clinic Study of Aging, a large community–based sample of people not selected for family history or risk of AD. At AAIC, Silke Kern of the Mayo and the University of Gothenburg, Sweden, reported MCSA NfL data from CSF, not blood. Kern showed longitudinal results in 648 participants. Their baseline NfL levels turned out to predict progression from normal to MCI better than their concentrations of CSF Aβ42, p-tau, T-tau, or the synaptic marker neurogranin.
Did NfL behave the same in blood? Michelle Mielke, also of the Mayo Clinic, reported on NfL concentrations in plasma from 79 people (64 cognitively normal, 15 with MCI) from the same study. She sent blood, collected at baseline and then again 15 or 30 months later, to be analyzed by Simoa in Kaj Blennow and Zetterberg’s group in Gothenburg. Baseline NfL did not correlate with baseline hippocampal volume, cortical thickness, amyloid PET, FDG-PET, or cognitive measures. However, higher baseline NfL did match with greater decline over time in the structural measures, FDG-PET and global cognition. A faster increase in NfL over the follow-up period signaled a steeper decline in global cognition. The effects were independent of amyloid levels.
The results mean plasma NfL could work as a short-term prognostic marker of ongoing neurodegeneration. The Mayo data, too, suggest that changes in plasma NfL parallel changes in cognition; however, Mielke needs larger samples and longer follow-up to say for sure. Like Weston, Mielke stressed the need to define what else affects plasma NfL in the general population.
In the proposed A/T/N framework for biomarker-based diagnosis and staging of AD, positive biomarkers for cerebral amyloid (A) and neurofibrillary tangles (T) define AD, while the neurodegeneration (N) category encompasses nonspecific measures of neuronal dysfunction and death. As proposed, this N category currently contains FDG-PET, brain atrophy, and CSF total tau (Apr 2018 news). “CSF NfL is a risk factor for cognitive decline, whether people are Aβ-positive or not. It’s not specific for AD, but might be a better marker for neurodegeneration in the A/T/N scheme,” said Kern. The results had many at the meeting asking if blood NfL might offer an even better measure of N.
In toto, while the older CSF-based NfL measurements are more established, every lab will now be trying the blood markers, Gordon told Alzforum. A combination of blood Aβ, which is specific for AD, and NfL, which is not, could revolutionize AD clinical trials and the ability to monitor people over time, he said. “Because blood NfL is a nonspecific marker, it should have high utility for other diseases, too,” he said.—Pat McCaffrey
With interest blooming in blood NfL as an easily accessed marker of neurodegeneration, disease severity, and prognosis (see Part 1 of this story), researchers are looking for practical applications. One study presented at the Alzheimer’s Association International Conference, held July 22–26 in Chicago, suggests that NfL could be used to stage candidate participants for early stage treatment trials. Another study is establishing NfL as a treatment marker in multiple sclerosis trials. Importantly, a study of cardiac surgery indicates that the protein could guide surgeons in developing more brain-sparing operating room techniques.
Holly Soares of AbbVie, Inc., North Chicago, Illinois, probed how plasma NfL tests might help sponsors screen for clinical trials in early AD as a way to shorten recruitment time and identify the people most likely to progress in the short term. In Soares’ experience, recruiting presymptomatic trial participants comes with a failure rate between 70 and 80 percent. For example, Bristol-Myer Squibb’s Phase 2 trial of avagacestat screened 1,358 people to enroll 287; A4 screened 4,486 people with amyloid PET to enroll 1,169 amyloid-positive cognitively normal participants (Aug 2018 conference news). Could blood T-tau or NfL more quickly or cheaply enrich for people likely to have brain amyloid deposition?
In a biomarker study, Soares recruited 25 people with AD, 45 with MCI, and 25 healthy controls who were all screened for amyloid positivity with CSF and PET. While CSF p-tau and T-tau were elevated in the amyloid-positive people, Soares detected no difference in plasma tau. Plasma NFL was elevated in AD and in people with MCI who were amyloid-positive, but not people with MCI who were amyloid-negative. However, the extensive overlap in absolute plasma NfL values between the groups suggested, to Soares’ mind, that NfL would not help screen for brain amyloid positivity.
On the other hand, plasma NfL was useful to enrich for fast progressors, a strategy to reduce the number of people needed to enroll in a clinical trial. Analyzing data from 574 ADNI participants, Soares compared baseline plasma NfL and total tau to cognitive decline on the CDR-sum of boxes over the following 90 months. People with MCI who were ApoE4 positive and had high plasma NfL and T-tau declined the fastest.
Those markers could reduce the necessary size of trials significantly, Soares showed. For example, a hypothetical study to detect a treatment effect in one or two years would require 159 people per arm based on existing recruitment protocols, but could run with 35 per arm by enrolling high NfL/high T-tau/ApoE4-positive participants with MCI. The numbers got even better when including Aβ information. “NfL is very promising,” Soares concluded. “It’s probably telling us who is in an active stage and likely to decline, while blood Aβ reflects underlying amyloid brain pathology.”
What does total tau signify? Soares said that’s “still an open question. The two are telling us different things, and we’ll need more than one blood biomarker to help enrich populations,” she said.
Another use of serum or plasma NfL might be to track treatment effects of neuroprotective drugs. Most of this work has been done in multiple sclerosis, and it offers promise for other diseases. In people with MS, serum NfL concentration correlates tightly with brain lesions and disease activity (Barro et al., 2018), and normalize after treatment (Disanto et al., 2017; Novakova et al., 2017). At AAIC, Charlotte Teunissen, VU University, Amsterdam, showed additional unpublished data from the Kuhle lab of a trial evaluating the MS drug fingolimod. In that trial, two years of treatment drastically decreased elevated plasma NfL, back down to the level of healthy subjects.
Hoping that this kind of success will translate to Alzheimer’s, researchers are now aiming to standardize blood NfL assays between labs. They are also trying to define both clinical reference values and what constitutes a meaningful change in patients. To do that, Teunissen described an ongoing, multisite analytical validation of the Simoa NfL assay in serum from MS patients. Scientists at 17 centers in North America and Europe will compare Simoa and ELISA according to a standardized protocol and common materials.
Teunissen stressed that variability in NfL between people, even in control groups, is an important issue scientists don’t yet understand. Age and gender contribute: “We see enormous variability by age, which leads to steady increase in NfL levels. We also see gender differences, which only emerge if you look at large cohorts,” she said.
What can AD researchers learn from MS? On the plus side, the magnitude of the rise in NfL is similar in MS and AD, Teunissen said. However, the MS field has highly effective treatments, and patients with the highest NfL levels show the strongest treatment response. “If NfL works best in patients with highest levels, we might be less optimistic that we can identify treatment effects in AD trials,” Teunissen said. Nonetheless, she noted, “Some AD patients have quite high NfL levels, so we may be able to see effects in that subset.”
In pediatric spinal muscular atrophy and neuropathy due to HIV infection, effective treatment also lowered NfL, according to data presented by Henrik Zetterberg, who works at both the University of Gothenburg and University College London. Seeing these dynamic changes gives Zetterberg hope for using NfL as a marker of treatment efficacy in AD. Zetterberg is so bullish on NfL that he told Alzforum, “I believe a drug against AD that does not reduce NfL has a low chance of being effective. Some would say I put too much faith in one biomarker, but time will tell.”
By conducting studies in boxers and other athletes, Kaj Blennow, University of Gothenburg, Sweden, has laid much of the groundwork for understanding how NfL rises and falls in response to acute brain injury. Now, he is turning his attention to probing the known link between heart surgery and postoperative cognitive decline. Surgical procedures are associated with delirium and dementia in some older patients, especially in people who start out with low CSF Aβ42, the marker for brain amyloid (Evered et al., 2016). Clinicians are not sure why. Could general anesthesia be damaging the brain?
Possibly so. Recently, Blennow and colleagues reported that surgical procedures in older patients cause an increase in plasma NfL and total tau (Evered et al., 2018). In the study, patients older than 60 who were undergoing major surgery saw their total tau peak at 2.5 times baseline six hours after surgery, and then start to decline; their NfL rose less, and more slowly, and was still at its maximum at 48 hours. Blennow said this is similar to the pattern seen in acute brain injury. The results suggest that general anesthesia during surgery may be associated with neuronal damage in the short term.
In the 2018 study, three out of four patients got hip or knee replacements. At AAIC, Blennow presented a follow-up study on 26 heart surgery patients, and reported that it had even more dire consequences. The surgery caused a spike in total tau that peaked at 20 times normal levels during surgery. NfL also spiked 20-fold, but did so a bit later, five days after surgery. Blennow detected no such a spike in 25 non-cardiac controls who underwent head or neck procedures.
NfL levels rose whether or not surgeons used a heart-lung machine to circulate blood, but they shot up significantly higher—20-fold versus eightfold—for patients on the pump than for those off the pump. These results indicate that unexpected variables may be putting the brain at particular risk during cardiac surgery. Above all, they suggest cardiac surgeons may be well advised to use blood biomarkers to optimize their procedures and to know how their patient’s brains are doing, Blennow said.
Do these surges in NfL and tau foreshadow postoperative cognitive decline? “That’s a key question,” Blennow said. “In our second study, we have acute cognitive assessment, and have preliminary results that there is a correlation. However, the real problems occur after three to six months, and we don’t have that data yet. We’ll collect it this year,” he said.
Attempts to publish this data in a journal read by surgeons have thus far been unsuccessful, Blennow added.—Pat McCaffrey
Technology for Patients: Purring Robots, Digital Data-Gathering
As technology becomes ever more part of modern life, researchers are exploring how it can help people with dementia. At a satellite conference held July 21 in Chicago, before the Alzheimer’s Association International Conference, speakers showcased the latest advancements in this field. Applications of technology fell into two broad categories: improving quality of life, and data-gathering and diagnosis.
Regarding the former, speakers claimed that robots can provide companionship for patients and a respite for caregivers, or even help patients with daily tasks. Most such robots remain in research use only, though one therapeutic model, the Paro baby harp seal, is commercially available and was on display at AAIC.
Regarding the latter, researchers said passive sensor technologies can gather more detailed behavioral data than traditional reports, and they presented digital diagnostic tests that appear to have greater sensitivity and real-world validity than paper-and-pencil methods. Data-gathering is more commercially advanced than robotics. Many devices and apps are available now, and trials are beginning to incorporate digital, continuous-outcome measures (May 2017 conference news).
Paro robot with its inventor, Takanori Shibata. [Courtesy of Gabrielle Strobel.]
“There’s been a profusion of technology in the areas of sensors, wearables, and mobile communications,” said Jeffrey Kaye of the Layton Aging and Alzheimer’s Disease Center at Oregon Health and Science University in Portland, Oregon. However, he noted that many technological applications remain mired at the pilot stage. Barriers to widespread adoption range from cost and insufficient evidence that they work, to rapid obsolescence and lack of standardization of many technical devices (Dec 2012 news series). Nonetheless, Alex Mihailidis of the University of Toronto told Alzforum that he expects technology for dementia patients to become more commercially available within the next five years.
Eva, Paro: Social Robotics Gain Acceptance
Perhaps no tech application generates as much fascination as robotics. These machines are still crude, a far cry from the sophisticated androids of film, but even simple machines could help people with dementia, speakers said. Dementia causes people to become isolated, depressed, agitated, and behaviorally challenged. Robots don’t run out of “patience,” and might make serviceable companions at later stages of the disease.
In Chicago, Jesus Favela of CICESE Research Center in Ensenada, Mexico, discussed a prototype social robot he calls Eva. Eva sits on a table, and somewhat resembles a diminutive, simplified R2-D2. A smart phone behind her? its? eyes lends Eva video capabilities, and a computer inside the body handles speech recognition. Favela designed Eva to deliver conversation, social stimulation, and distraction for users, as well as potentially to intervene with problem behaviors.
Favela and doctoral student Dagoberto Cruz tested Eva in a pilot study of 12 dementia patients, whose average age was 80 and MMSE 14. In initial sessions with the participants, Eva asked questions about what foods and music they liked, and noted their conversation style. The researchers used the information to develop a personalized profile for each participant, which facilitated future interactions. For example, Eva would pause longer when talking with chatty people to give them time to speak. In these sessions, Eva often entertained small groups of participants by playing their favorite music. Favela showed a video of Eva playing a song and suggesting the listeners stand up and dance, which they did. In later sessions, Eva initiated more free-ranging conversations, or engaged participants by asking them to complete popular sayings.
Eva also helped moderate behavior. In another video, an agitated patient got up and walked away from the session. Eva asked her to please stay, and the woman returned, telling her companion, “She asked me so nicely, I have to stay.” In some sessions, Eva led relaxation exercises, suggesting her listeners close their eyes and breathe slowly.
Participants with dementia talk with Eva. [Courtesy of Jesus Favela.]
Perhaps surprisingly to an audience of cognitively normal, naturally skeptical scientists, the participants in this small study appeared to accept the robot as a conversation partner. Favela said they remembered Eva from one session to the next, although they did not always remember its name, or when they last interacted with it. Participants interacted, smiled, and laughed more as sessions went on, Favela added.
Sometimes conversations broke down. For example, when several people spoke simultaneously, Eva could not follow. However, participants tolerated these lapses and did most of the work of getting the conversation back on track, Favela said.
Participants did not seem to notice Eva’s digital nature—or did they tease Eva? In one video, a group of women told Eva their favorite food was mangos. When the robot said it did not eat mango, one woman demanded, “Why would you not eat such a delicious food?” “Because I am a robot,” Eva replied, sparking gales of laughter. Favela told Alzforum that the participants seemed to shift between treating the robot as an object and giving it a personality. One woman often invited Eva to visit her hometown, promising to host the robot at her house and cook meals for it, Favela said.
Favela and Cruz are working on upgrading Eva’s capabilities to allow it to read physiological signs, such as heart rate and temperature, and to adapt to the mood of its conversation partners. He believes the robot could be programmed to perform simple behavioral interventions, such as telling a person who wakes up in the middle of the night that it is not time to get up. Favela hopes Eva will be commercially available in the future.
While Eva is evolving, the Paro robot is already in widespread commercial use. Takanori Shibata of the National Institute of Advanced Industrial Science and Technology in Tsukuba, Japan, spearheaded its development in 2002. Shibata told Alzforum he designed Paro to look like a harp seal because people have few expectations about how this animal might behave, and therefore accept the robot more readily than they would a mechanical dog or cat. In Chicago, a handful of Paros stuck out their noses and soft, furry heads on a high-traffic corner booth of the exhibit hall, and few who passed by it could resist petting it. Paro blinks its eyes, moves in response to touch, and chirps and purrs. When cuddled, it wraps its flippers around its handler and rests its head on his or her shoulder. Paro’s software allows it to modify its behavior in response to a person’s actions, for example learning what elicits positive interactions such as petting, and then repeating that behavior.
Paro is now in its ninth generation, and has been in use in Japan and Europe since 2003. The company offers three different versions: The white seal is the most interactive and popular model, the gray seal is sleepy and snuggly, and the tan seal moves slowly. Paro’s price tag is $6,400.
A number of small studies have found that interacting with Paro benefits dementia patients. In a randomized controlled trial, those who played with Paro reported feeling less lonely, and in a pilot study, nursing home residents with access to Paro appeared calmer and happier, with fewer instances of yelling, pacing, and anxious behavior (Robinson et al., 2013; Lane et al., 2016). Other studies report that treatment groups score lower on measures of stress and anxiety, and have lower blood pressure after interacting with Paro. In randomized controlled trials, treatment groups used less psychotropic and pain medicine than controls (Robinson et al., 2015; Jøranson et al., 2016; Petersen et al., 2017). In 2009, the U.S. Food and Drug Administration approved Paro as a class II neurological therapeutic device.
When Will Robot Aides Be Feasible?
Designing a robot that renders more than soothing pet services remains a formidable challenge, and some groups are working on it. For example, François Michaud of the University of Sherbrooke, Canada, envisions robots that monitor people with dementia at home and report problems to family.
Michaud first adapted Amazon’s Beam Plus telecommunications device, a monitor on wheels designed for audio and video phone calls. Besides adding computing power and sensors, Michaud also enabled the device to move around a room and return to its docking station as needed, using a program called Simultaneous Localization and Mapping. His robot has speech-recognition software and tracks faces and voices. It can monitor vital signs such as heart rate and temperature (Lepage et al., 2016; Laniel et al., 2017).
Michaud is currently testing whether this robot could monitor people with dementia at night. He is working to improve its software by adding the ability to recognize common actions and to infer what task a person might be trying to accomplish, for example making a cup of tea. Ideally, the robot could then suggest tips. Michaud will build in spatiotemporal episodic memory so the robot can learn from human behavior patterns. Robots need to have contextual awareness in order to engage in meaningful interaction with people, Michaud noted. His is at the prototype stage; its base technology runs at $2,000.
In Chicago, speakers discussed more challenges they need to overcome. For one thing, a person’s speech changes as his or her dementia progresses, making it more difficult for software to interpret. Autonomous robots raise potential ethical concerns—could they make decisions for their owners, taking away their agency? Mobile robots could fall over and hurt people. Even so, researchers at AAIC thought robotic aides hold promise, with a favorable cost/benefit ratio for people with dementia.
Stream of Continuous Data From Passive Monitoring
Unlike the field of robotics, tech monitoring is no longer in its infancy. Its goal is to collect information in the background, through seamless, invisible applications. “The most effective technology is the most passive,” Kaye noted. One such system is Emerald, developed at MIT. From a small white box mounted to the wall, Emerald emits radio waves and detects their reflection from nearby objects to map its environment. It uploads raw data to a server and uses machine-learning algorithms to extract information about a person’s movements, location, and sleep patterns. It can measure respiration based on the movement of a person’s chest.
Daily Log.
Color-coded sensor data show how much time a 75-year-old study participant with vascular dementia, apathy, sleepiness, and anxiety spent in each activity. Circle represents 24 hours, concentric rings are separate days. [Courtesy of Ipsit Vahia.]
In a pilot study of how well Emerald gathers data on daily activities, Ipsit Vahia of McLean Hospital in Belmont, Massachusetts, installed it in the rooms of three volunteers in an assisted living center for four months. Emerald tracked pacing, walking speed, time spent in bed or in a chair, and sleep quality. Compared with observations by facility staff, Emerald generated more detailed data. Distinct patterns for each participant emerged, Vahia said at AAIC. Emerald identified sleep apnea, late-night waking and, in one case, showed that a participant became more agitated after having visitors. The data suggests Emerald could indeed spot behavioral changes and identify triggers for problem behavior, Vahia said.
But would nurses and aides use such data? Perhaps not so much. Katherine Wild, also at the Layton Aging and Alzheimer’s Disease Center, examined how well a similar passive monitoring system worked in retirement communities. Ninety-five residents in seven facilities participated for about two years each, and center staff could access the data via a computerized “dashboard.” The idea was that this might help caregivers decide when residents needed to move to a higher level of care. Alas, staff rarely logged in and did not use the data for decision-making, even though they had been involved in developing the dashboard. They told researchers there was too much data, and that it was difficult to differentiate acute events from trends. Some had trouble logging into the system, or had ethical qualms that viewing the data violated patient privacy. Overall, the technology was not sufficiently user-friendly, Wild concluded.
Virtual View. A VR maze combines the skills of real-world navigation and map-reading. [Courtesy of Raquel da Costa.]
Diagnostic Applications Gain Traction
Can technology improve diagnosis? Some digital measures are starting to trump older tests, speakers at AAIC agreed. For example, Raquel da Costa of the University of São Paulo noted that people with prodromal AD have trouble navigating through environments (Oct 2015 news; Jan 2017 news). Traditionally, researchers assess this deficit with the MRMT, aka Money Road-Map Test of Direction Sense, a paper-and-pencil test. However, looking at a two-dimensional map bears little resemblance to real-world navigating, so da Costa and colleagues wondered if a three-dimensional, virtual-reality maze would work better. Translating the MRMT into a VR experience, the research group developed the Spatial Orientation in an Immersive Virtual Environment Test (SOIVET) (da Costa et al., 2018).
To compare the two, da Costa enrolled 30 cognitively healthy people aged 18 to 59. Each completed the Santa Barbara Sense of Direction Scale survey to measure their own perception of how well they navigated. Then they took the MRMT as well as the virtual-reality version. For the latter, participants wore a headset giving them an immersive first-person view of a virtual world (see image). At their feet, they saw a two-dimensional map of their surroundings and used this information to find their way. In this way, the virtual-reality test mimicked real-world map-reading, requiring participants to split their attention between two tasks, da Costa said.
Participants found the virtual-reality task more difficult than the paper version, averaging 13 correct responses on the former versus 30 correct responses on the latter. Performance on the two tasks correlated with each other, suggested they measured the same skill. However, only the VR test score correlated with participants’ own sense of how well they could navigate; the MRMT did not correlate to the survey results. This suggests that the VR task better recreates real-world navigation and assesses spatial abilities more like those used in real life, da Costa said.
Other diagnostic tests use drawing to assess decline. A digital version of the clock-drawing task is already known to beat the paper-and-pen version (Aug 2018 conference news), and Kelvin Tsoi of the JC School of Public Health and Primary Care in Shatin, Hong Kong, described a different drawing test for which this appears to be true, as well. Noting that some dementia screening tests ask people to draw geometric shapes, Tsoi asked participants to draw two interlocking pentagons on a digital tablet while looking at a reference figure. Like the digital pen, the tablet measured factors such as drawing speed, hesitation, and stops at corners.
The researchers compared the performances of 194 AD patients seen at Hong Kong clinics to those of 271 cognitively healthy elderly recruited from community centers. The patients had an average age of 80, controls, 76. The former took an average 17.5 seconds to draw the shapes, compared with 12.6 seconds for controls. A machine learning algorithm predicted whether each participant had AD, based on his or her performance, and “diagnosed” AD with a sensitivity of 78 percent and specificity of 71 percent. Tsoi said that this beats the MMSE, which in this cohort had a sensitivity of 73 percent and specificity of 53 percent.
These talks but scratched the surface of technology research in AD. Other speakers noted advances in wearable technology, apps, and the design of “smart homes.” A key ingredient in taking these applications from research to real world will be to involve people with dementia in their design, speakers agreed.—Madolyn Bowman Rogers

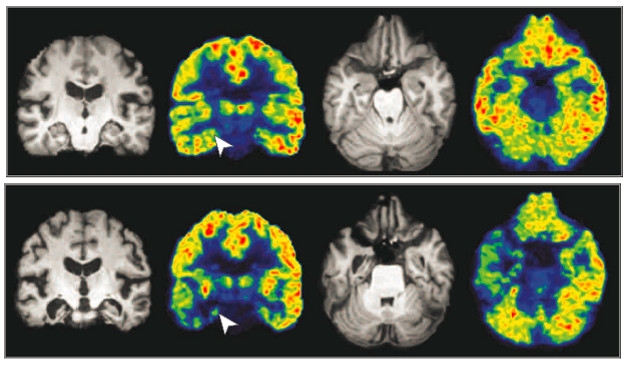





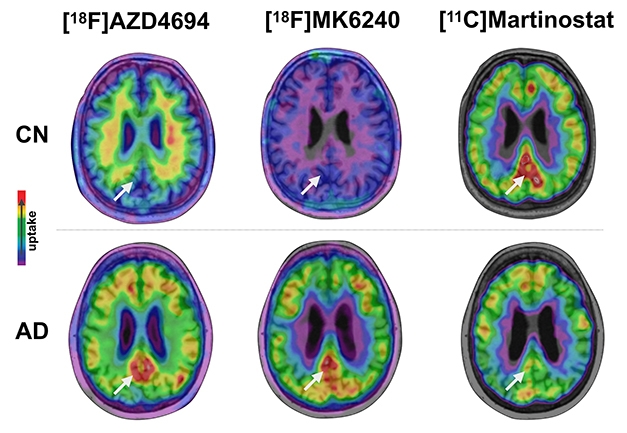




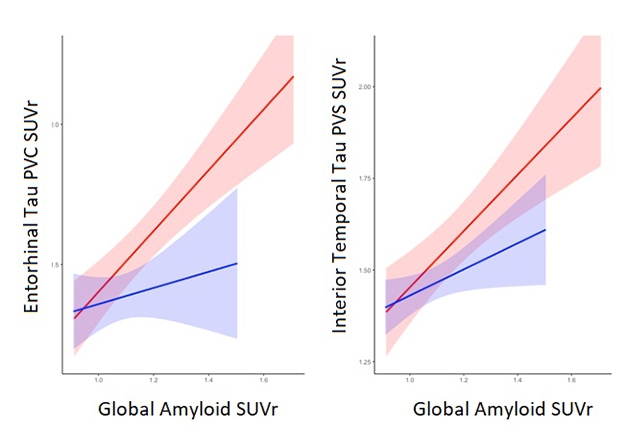







Comments
No Available Comments
Make a Comment
To make a comment you must login or register.