Meta-Analyses Deliver Most Definitive Data Yet on Amyloid Prevalence
Quick Links
Two meta-analyses offer up the largest data sets to date on how commonly amyloid builds up in people’s brains. Published May 19 in JAMA and led by Pieter Jelle Visser at VU University Medical Center in Amsterdam and Maastricht University, both in The Netherlands, the two studies compiled amyloid PET and cerebrospinal fluid (CSF) biomarker data from thousands of participants. One meta-analysis looked at the prevalence of amyloid in cognitively normal people, and concluded that amyloid creeps into the brain 20 to 30 years before dementia can be diagnosed. This was particularly true for people who carry an ApoE4 allele; indeed, they developed amyloid at a younger age. In their second study, Visser and colleagues compared amyloid prevalence among people clinically diagnosed with AD or other dementias, including dementia with Lewy bodies, frontotemporal dementia, and corticobasal syndrome. They found that the prevalence of brain amyloid in people diagnosed with most non-AD dementias was higher with increasing age. They concluded that older people may be likelier to have multiple pathologies, or to have been misdiagnosed. The data may help researchers set inclusion criteria for clinical trials, or make better diagnoses.
“These are outstanding meta-analyses,” commented Michael Weiner of the University of California, San Francisco. The lag between the appearance of amyloid and the onset of dementia is a bit longer than previous estimates, Weiner wrote. He also cited limits on interpreting the results—such as the study’s cross-sectional design and the idea that people with high cognitive reserve may stave off dementia longer than others. “The final answer will come from large longitudinal studies,” he wrote.
Knowing the prevalence of amyloid pathology in people without dementia is crucial for designing AD prevention trials. Anywhere from 10 to 60 percent of people without dementia harbor amyloid plaques upon autopsy, and studies that rely on biomarkers, such as amyloid PET or CSF Aβ42, as a proxy for amyloid are equally variable (see Murayama et al., 2004; Bennett et al., 2005; Lin et al., 2009; Rowe et al., 2010; Randall et al., 2013). Another problem that dogs trial design is misdiagnosis and/or mixed pathology. Some people diagnosed with AD have turned out to harbor no amyloid in the brain, whereas some others diagnosed with a non-AD dementia do have underlying amyloid pathology that could be a factor in their disease. Moreover, this issue can make it difficult for a physician to choose the best course of treatment for a given patient.
Visser sought to clarify and bring consensus to these issues by performing the two meta-analyses. To assess the prevalence of amyloid positivity in people without dementia, first author Willemijn Jansen of Maastricht University, The Netherlands, and colleagues scoured the literature for studies that assessed amyloid PET and/or CSF Aβ42, and ApoE status. They identified 91 papers; of these, the authors of 54 agreed to share participant-level data, while another shared group-level data. These 55 papers included 45 single-center and 10 multicenter cohorts. About half of the studies used amyloid-PET and half CSF Aβ42. They included 7,583 participants, of whom 2,914 were cognitively normal, 697 had subjective cognitive impairment (SCI), and 3,972 were diagnosed with mild cognitive impairment (MCI).
In people with normal cognition, the prevalence of amyloid increased with age, from 10 percent in 50-year-olds to 23 percent in 70-year-olds to 43 percent in nonagenarians. The prevalence was similar among people with SCI, but double that in the MCI group. This suggests that MCI, though defined somewhat differently across reports in the meta-analysis, represents a true stage on the AD spectrum, while SCI does not, the authors concluded.
In general, the prevalence of amyloid was two to three times greater in ApoE4 carriers than non-carriers. The exact number of this increased prevalence depended on their age and cognitive diagnosis. The gap between ApoE4 carriers and non-carriers widened with age, and was similar among people with normal cognition, SCI, or MCI. Age-related prevalence differed across ApoE genotypes. For example, the age at which 15 percent of participants with normal cognition tested positive for amyloid was 40 years for E4/E4 carriers, and then steadily increased among E2/E4, E3/E4, E3/E3, and E2/E3 carriers, who were over 85 (see figure below). None of the 10 E2/E2 carriers included in the analysis had amyloid. While the E2 allele fended off amyloid, this protection crumbled when the E4 allele was also present, as the difference in amyloid prevalence between E2/E4 and E3/E4 carriers was not significant. Among 90-year-olds with normal cognition, 40 percent of ApoE4 non-carriers and 80 percent of carriers had amyloid. The researchers obtained similar results from studies that used amyloid PET or CSF Aβ42.

ApoE: the 600-Pound Gorilla? People carrying the ApoE4 allele were more likely to become amyloid positive earlier in life. Shading indicates 95 percent confidence interval for each curve. [Image courtesy of Jansen et al., copyright 2015 American Medical Association.]
Years of education also affected amyloid. Its prevalence was 5 percent higher among cognitively normal 70-year-olds with above-average education than among their less-educated peers. This result agrees with the idea of cognitive reserve, the researchers said, because education could protect people from the damaging effects of amyloid pathology, allowing them to remain cognitively normal with a higher amyloid burden than less-educated people. Finally, the researchers compared the prevalence of amyloid with that of Alzheimer’s dementia. As has been reported by the Australian Imaging, Biomarkers, and Lifestyle (AIBL) study, the Dutch meta-analysis also found that amyloid precedes dementia by about 20 years. In other words, if the prevalence of amyloid positivity is 30 percent at age X in a given population, then that same population’s prevalence of dementia is 30 percent at age X+20 years.
“This study is the most definitive statement to date on the prevalence of amyloid and how it relates to ApoE,” commented Christopher Rowe of the University of Melbourne in Australia. He said the large numbers of participants in the studies bring more certainty to previously reported findings. “The information is important, not only for researchers, but for identifying appropriate cohorts for clinical trials,” said Rowe, who contributed amyloid PET data from more than 200 people in AIBL to the meta-analysis.
In the second paper, first author Rik Ossenkoppele of VU University Medical Center in Amsterdam meta-analyzed rates of amyloid positivity in people with AD or other dementias. They searched the literature for studies that assessed amyloid PET, ApoE genotype, and listed dementia diagnoses that were based on clinical criteria, rather than the results of the PET scan. In all, they analyzed 185 articles on 29 cohorts of patients. Of the 1,897 patients, 1,359 were clinically diagnosed with AD, 288 with frontotemporal dementia (FTD), 138 with vascular dementia (VaD), 51 with dementia with Lewy bodies (DLB), and 61 with corticobasal syndrome (CBS). As reference populations, the researchers also incorporated data on 1,849 healthy controls, and 1,369 patients with confirmed AD at autopsy.
People diagnosed with AD had the highest prevalence of amyloid—88 percent. Interestingly, prevalence was higher in younger people, ranging from 93 percent in 50-year-olds to 79 percent in 90-year-olds. The likeliest explanation for this trend, Visser speculated, is misdiagnosis. The prevalence of other pathologies that mimic AD, such as hippocampal sclerosis, increases with age, he noted. This means older people may be more likely to be misdiagnosed with AD than younger people who are at a lower risk for these other morbidities. Claudia Kawas of the University of California, Irvine, agreed with that hypothesis. She added that despite the low numbers of very old people included in the studies, the results agreed with observations from the 90+ Study of the oldest old: “Amyloid positivity increases with age and with ApoE4, but its relationship with dementia diminishes with age,” Kawas said. Another possible explanation is that some older people had low levels of amyloid pathology that were not detected on the scans, yet due to their age were enough to cause dementia, Visser offered.
Clifford Jack of the Mayo Clinic in Rochester, Minnesota, who was not involved in the study, melded these two possibilities together. He suggested other pathologies make older people more susceptible to the effects of amyloid. “Young people need a lot of AD pathology to become demented because they have few other pathologies driving down cognition,” he wrote. “An older person with a little amyloid (i.e., AD pathology) can become demented because of the presence of additional age-related pathologies.”
Among ApoE4 carriers diagnosed with AD, amyloid prevalence was consistently 90 percent, regardless of age. Autopsy data on AD patients revealed similar rates of amyloid positivity as the PET scans did.
Amyloid varied considerably across non-AD dementias. For example, 51 percent of people diagnosed with DLB harbored amyloid, as did 30 percent of people with VaD, 12 percent of people with FTD, 38 percent of people with CBS, and 22 percent of healthy controls. Therefore, all dementias except FTD had higher amyloid prevalence than healthy controls did. In contrast to the trend seen in AD patients, people with DLB, VaD, or FTD had increasing amyloid prevalence with age (see figure below). Corticobasal syndrome was the exception; like people with AD, older CBS patients were less likely to have amyloid. Across all syndromes, amyloid prevalence was higher in ApoE4 carriers than non-carriers.
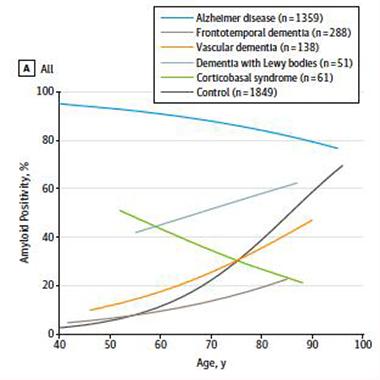
Amyloid Convergence.
Amyloid prevalence declines with age in people diagnosed with AD or CBS, but increases with age in people with FTD, VaD, DLB, and in healthy controls. [Image courtesy of Ossenkoppele et al., copyright 2015 American Medical Association.]
What does amyloid in people with non-AD dementias mean? They may have been misdiagnosed and had AD, or they may have had AD pathology in addition to the dominant pathology that drove their other diagnosis. While either of these possibilities could be true for a given person, Visser speculated that mixed pathology is more likely. While amyloid prevalence increases with age in people with non-AD dementias just as it does in cognitively normal people, he believes another disease-specific pathology, such as tau or TDP-43, could dictate the course of their diseases and diagnoses. This is not to say that amyloid does not affect those with non-AD dementia—in fact, the presence of amyloid correlated with lower MMSE scores in these groups. People with non-AD dementia who also harbor amyloid could have AD or prodromal AD in addition to their other condition, Visser said.
In the case of CBS, Visser said the underlying pathology of the disease is highly heterogeneous. At autopsy, amyloid plaques were revealed as the primary driver of the disease in 25 percent of patients (see Dickson et al., 2002; Lee et al., 2011). Visser speculated that amyloid drives the disease in younger people diagnosed with CBS, while tau predominates in the older population. This will become clear as tau tracers become available, he said.
David Holtzman of Washington University in St. Louis, who was not involved in the study, offered similar explanations for the trends. “Amyloid deposition gets more common the older one is after age 50 in cognitively normal people, so it is not surprising it gets more common in the setting of these other diseases with aging,” he wrote to Alzforum. “In corticobasal syndrome, the syndrome may be due to AD. In the case of FTD and vascular dementia, it may be that many of these people are developing preclinical AD, which gets more common the older people are.” (See full comment below.)
What does this data mean for the use of PET scans in diagnosis? It more sharply defines its usefulness in certain subgroups. For example, the rise in amyloid positivity with age among most non-AD dementias indicates that amyloid PET scans may be less useful for differential diagnosis at older ages, Visser said. “In elderly subjects, the ability of amyloid positivity to differentiate between AD and other conditions becomes weak,” he said. In the case of ApoE4 carriers with a diagnosis of AD, PET scans may be unnecessary to confirm a diagnosis, given that 90 percent of them will have amyloid regardless of their age, he added.
“The value of these observations is that amyloid imaging may be most critical in making the correct diagnosis in early onset dementia, especially to rule in AD dementia,” wrote Roger Rosenberg from the University of Texas Southwestern Medical Center at Dallas in an accompanying editorial in JAMA. He added that the data will aid in the design of prevention trials.—Jessica Shugart
References
Paper Citations
- Murayama S, Saito Y. Neuropathological diagnostic criteria for Alzheimer's disease. Neuropathology. 2004 Sep;24(3):254-60. PubMed.
- Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005 Mar 8;64(5):834-41. PubMed.
- Lin YT, Cheng JT, Yao YC, Juo, Lo YK, Lin CH, Ger LP, Lu PJ. Increased total TAU but not amyloid-beta(42) in cerebrospinal fluid correlates with short-term memory impairment in Alzheimer's disease. J Alzheimers Dis. 2009;18(4):907-18. PubMed.
- Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, Fripp J, Tochon-Danguy H, Morandeau L, O'Keefe G, Price R, Raniga P, Robins P, Acosta O, Lenzo N, Szoeke C, Salvado O, Head R, Martins R, Masters CL, Ames D, Villemagne VL. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010 Aug;31(8):1275-83. PubMed.
- Randall C, Mosconi L, de Leon M, Glodzik L. Cerebrospinal fluid biomarkers of Alzheimer's disease in healthy elderly. Front Biosci (Landmark Ed). 2013;18:1150-73. PubMed.
- Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I, . Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002 Nov;61(11):935-46. PubMed.
- Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, Dearmond SJ, Huang EJ, Trojanowski JQ, Growdon ME, Jang JY, Sidhu M, See TM, Karydas AM, Gorno-Tempini ML, Boxer AL, Weiner MW, Geschwind MD, Rankin KP, Miller BL. Clinicopathological correlations in corticobasal degeneration. Ann Neurol. 2011 Aug;70(2):327-40. PubMed.
Further Reading
Papers
- Klunk WE. Amyloid imaging as a biomarker for cerebral β-amyloidosis and risk prediction for Alzheimer dementia. Neurobiol Aging. 2011 Dec;32 Suppl 1:S20-36. PubMed.
- Chételat G, La Joie R, Villain N, Perrotin A, de La Sayette V, Eustache F, Vandenberghe R. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer's disease. Neuroimage Clin. 2013;2:356-65. Epub 2013 Mar 5 PubMed.
Primary Papers
- Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Amyloid Biomarker Study Group, Aalten P, Aarsland D, Alcolea D, Alexander M, Almdahl IS, Arnold SE, Baldeiras I, Barthel H, van Berckel BN, Bibeau K, Blennow K, Brooks DJ, van Buchem MA, Camus V, Cavedo E, Chen K, Chetelat G, Cohen AD, Drzezga A, Engelborghs S, Fagan AM, Fladby T, Fleisher AS, van der Flier WM, Ford L, Förster S, Fortea J, Foskett N, Frederiksen KS, Freund-Levi Y, Frisoni GB, Froelich L, Gabryelewicz T, Gill KD, Gkatzima O, Gómez-Tortosa E, Gordon MF, Grimmer T, Hampel H, Hausner L, Hellwig S, Herukka SK, Hildebrandt H, Ishihara L, Ivanoiu A, Jagust WJ, Johannsen P, Kandimalla R, Kapaki E, Klimkowicz-Mrowiec A, Klunk WE, Köhler S, Koglin N, Kornhuber J, Kramberger MG, Van Laere K, Landau SM, Lee DY, de Leon M, Lisetti V, Lleó A, Madsen K, Maier W, Marcusson J, Mattsson N, de Mendonça A, Meulenbroek O, Meyer PT, Mintun MA, Mok V, Molinuevo JL, Møllergård HM, Morris JC, Mroczko B, Van der Mussele S, Na DL, Newberg A, Nordberg A, Nordlund A, Novak GP, Paraskevas GP, Parnetti L, Perera G, Peters O, Popp J, Prabhakar S, Rabinovici GD, Ramakers IH, Rami L, Resende de Oliveira C, Rinne JO, Rodrigue KM, Rodríguez-Rodríguez E, Roe CM, Rot U, Rowe CC, Rüther E, Sabri O, Sanchez-Juan P, Santana I, Sarazin M, Schröder J, Schütte C, Seo SW, Soetewey F, Soininen H, Spiru L, Struyfs H, Teunissen CE, Tsolaki M, Vandenberghe R, Verbeek MM, Villemagne VL, Vos SJ, van Waalwijk van Doorn LJ, Waldemar G, Wallin A, Wallin ÅK, Wiltfang J, Wolk DA, Zboch M, Zetterberg H. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015 May 19;313(19):1924-38. PubMed.
- Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, van der Flier WM, van Berckel BN, Scheltens P, Visser PJ, Amyloid PET Study Group, Verfaillie SC, Zwan MD, Adriaanse SM, Lammertsma AA, Barkhof F, Jagust WJ, Miller BL, Rosen HJ, Landau SM, Villemagne VL, Rowe CC, Lee DY, Na DL, Seo SW, Sarazin M, Roe CM, Sabri O, Barthel H, Koglin N, Hodges J, Leyton CE, Vandenberghe R, van Laere K, Drzezga A, Forster S, Grimmer T, Sánchez-Juan P, Carril JM, Mok V, Camus V, Klunk WE, Cohen AD, Meyer PT, Hellwig S, Newberg A, Frederiksen KS, Fleisher AS, Mintun MA, Wolk DA, Nordberg A, Rinne JO, Chételat G, Lleo A, Blesa R, Fortea J, Madsen K, Rodrigue KM, Brooks DJ. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA. 2015 May 19;313(19):1939-49. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
San Francisco Veterans Affairs Medical Center
My overall comment about both papers is that these are outstanding meta-analyses, which pull together all of the published data concerning the prevalence of amyloid positivity in various dementias, in normal and MCI subjects, and the effects of age and APOE genotype. The paper by Jansen et al. concerning the prevalence of amyloid positivity in the cognitively normal and MCI cases concluded in the abstract that “[t]hese findings suggest a 20- to 30-year interval between first development of amyloid positivity and onset of dementia.” This conclusion is likely to draw notice in both the scientific community and in the public press.
In their discussion sections, the authors review several caveats about making such conclusions, including that this is a cross-sectional study, self-selection bias, and the possibility that “cognitive reserve” may allow some subjects to resist the downstream effects of amyloid pathology. Other investigators using data from other sources have estimated that there might be only a 15-year window between development of significant amyloid pathology and onset of dementia. The final answer will come from large longitudinal studies of normal subjects, MCI, and patients with AD that use amyloid phenotyping and cognitive follow-up. The Alzheimer’s Disease Neuroimaging Initiative is one example of this approach.
We can all agree that many years elapse between the development of significant amyloid pathology, which is detected by amyloid PET scans or CSF measurements of amyloid beta, and the onset of cognitive decline leading to dementia.
Furthermore, our field has reached consensus that AD is an “amyloid- facilitated tauopathy,” in which the accumulation of tau tangles is more closely correlated with synapse loss, neurodegeneration, and the development of cognitive decline than the accumulation of amyloid beta. Therefore, there is a substantial window for “prevention trials” using lifestyle modifications or pharmaceutical treatments to prevent the symptoms and disability due to AD.
Washington University
Regarding Jansen et al., this paper estimates the percentage of people who are amyloid positive from a cross-sectional meta-analysis of published studies in cognitively normal people, people with subjective cognitive impairment, and people with mild cognitive impairment. The findings are similar to what has been suggested from many previous studies, i.e., that amyloid deposition begins in some people starting after age 40 and that the percentage of amyloid-positive people is greatly dependent on ApoE genotype.
Other studies have estimated that amyloid deposition begins about 10-20 years prior to the onset of symptomatic Alzheimer’s disease or very mild dementia due to AD. The authors here argue that amyloid deposition may begin 20-30 years prior to the onset of cognitive decline due to AD. I think longitudinal studies that are ongoing will be required to determine the timeframe following amyloid deposition in which cognitive decline begins to occur.
Ossenkoppele et al. looked at the percentage of people who are amyloid positive with a clinical diagnosis of AD as a function of age as well as the percentage of people who have different neurodegenerative diseases such as FTD, corticobasal syndrome, and vascular dementia (as well as cognitively normal people) as a function of age. They found that for individuals diagnosed with AD in their 50s, more than 90 percent were amyloid positive whereas by around age 90, a little less than 80 percent were amyloid positive. This is consistent with the fact that as people get older, a number of additional age-related diseases can mimic or phenocopy AD (hippocampal sclerosis, tangle predominant dementia, vascular dementia).
They also found that in FTD, vascular dementia, and dementia with Lewy bodies, the percentage of amyloid-positive people with these disorders increased with age (only studied until about age 80). Amyloid deposition gets more common the older one is after age 50 in cognitively normal people, so is not surprising it gets more common in the setting of these other diseases with aging. In some cases, in particular corticobasal syndrome, the individuals may have their corticobasal syndrome from AD. In the case of FTD and vascular dementia, it may be that many of these people are developing preclinical AD, which gets more common the older people are.
Case Western Reserve University
The meta-analysis presented in these two papers should be very useful for AD researchers to test their favorite hypotheses of AD pathogenesis. Although most of the observations reported here are not surprising, one finding that stands out is the awareness that amyloid deposition begins even earlier—20-30 years prior to the onset of clinical symptoms—than the 10-15 year period previously thought. However, this finding should be not be surprising either, because Braak and colleagues (Braak et al., 2011), based on 2,332 brain autopsies from individuals 0 to 100 years old on, had previously found that amyloid deposits begin as early as 40 years of age and are found in 75-80 percent of all brains after the age of 80 years. (They also found that tau pathology begins in a different region of the brain and at least a decade before amyloid deposition occurs, but this finding does not receive much attention.)
There are many interesting findings in this study. I would like to comment only on the real but poor correlation between amyloid deposits and AD and the “illusion of causality” (Yarritu and Matute, 2015) that it confers. This study shows that amyloid positivity increases with age in different populations examined (cognitively normal, SCI, MCI, or patients with dementia syndromes) and the rate is remarkably similar (the curves are approximately parallel). By contrast, the rate actually drops with age in the AD population. They also reported increased amyloid positivity with higher education levels, which paradoxically protect from developing AD. (The concept of “cognitive reserve” has been proposed to explain this paradox, but it is a circular argument. Plus, it is difficult to imagine Nobel laureate scientists and writers with AD as having low cognitive reserve.)
The findings from these two papers can be interpreted as supporting the amyloid hypothesis because of the higher prevalence of amyloid positivity in MCI/AD. However, one would essentially find a similar picture even if AD and amyloid positivity were independent of each other, and were caused by aging—a scenario similar to yellow teeth and lung cancer being independent but correlated with and caused by smoking. The causality of amyloid in AD can only be proven by a robustly successful anti-amyloid clinical trial. Should the currently ongoing secondary prevention trials also fail, then the question will remain open as to whether they failed because the amyloid pathology starts even earlier than we thought (and even the presymptomatic intervention came too late) or because the amyloid hypothesis is flawed, as many of us have argued.
References:
Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011 Nov;70(11):960-9. PubMed.
Yarritu I, Matute H. Previous knowledge can induce an illusion of causality through actively biasing behavior. Front Psychol. 2015;6:389. Epub 2015 Apr 8 PubMed.
Make a Comment
To make a comment you must login or register.