Estrogen-like Receptor: The Gas Pedal of the Brain?
Quick Links
Brains are high-maintenance. All that thinking accounts for a whopping 25 percent of the glucose a body burns, and 20 percent of the oxygen it consumes, yet the stuff inside the noggin only weighs about three pounds—a mere 2 percent of the body's mass. How do brain cells rev up their metabolism to meet the energy demands? With the help of estrogen-related receptor gamma (ERRγ), according to a paper in the April 7 Cell Metabolism. Scientists led by Liming Pei, Perelman School of Medicine, University of Pennsylvania, Philadelphia, and senior author Ronald Evans, Salk Institute for Biological Studies, La Jolla, California, found that the nuclear receptor, which turns on metabolic genes, helps provide the energy that powers long-term potentiation, learning, and memory. Mice lacking the receptor have trouble navigating a water maze.
“This is an important finding,” said M. Flint Beal of Weill Cornell Medical College, New York, who was not involved in the research. “It suggests that if you could boost energy metabolism with a small molecule that activates ERRγ, you might be able to compensate for some of the bioenergetic defects that occur in diseases such as Parkinson’s and Alzheimer’s, and perhaps improve memory.”
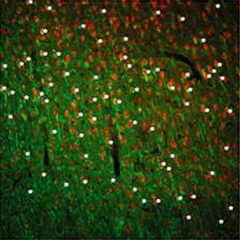
To ERRγ Is Right for Neurons.
White asterisks mark neurons (green) expressing ERRγ (red). [Image courtesy of Pei et al., 2015, Cell Metabolism.]
Neurons burn glucose for energy. They shuttle metabolites of glycolysis into the tricarboxylic acid (TCA) cycle and oxidative phosphorylation pathways that generate ATP. Glucose and oxygen consumption are tightly coupled to brain activity. The more a neuron fires, the more fuel it needs to feed its ATP appetite. What underlies this coupling? While previous studies suggested that neurons switch on specific sets of genes when most active, the transcription factors responsible remain unclear (Alberini, 2009; Magistretti, 2006).
To identify those key transcription factors, Pei and colleagues examined mouse embryonic stem cells that were just differentiating into neurons. At this point, the cells ramp up their energy output and the production of mitochondria. Pei examined the fledgling neurons for an uptick in transcription factors that were known to regulate cell metabolism in other cells. Levels of one—ERRγ—rose 100-fold in the differentiating neurons. In the adult mouse, ERRγ was widely expressed in most neurons and some astrocytes in various brain regions, including the cerebral cortex, hippocampus, and dentate gyrus (see image above).
Could ERRγ drive up metabolism? Using a variety of genomic methods, Pei and colleagues found that in the differentiating neurons, ERRγ crowded promoters, intergenic regions, and introns of hundreds of genes, much like it does in the periphery. Most of the genes are involved in ATP generation, oxidative phosphorylation, glycolysis, the TCA cycle, and mitochondrial function. ERRγ did not bind genes involved in fatty acid breakdown, as it does in heart cells (Dufour et al., 2007). When the researchers knocked out ERRγ in mice, neurons in the cerebral cortex expressed fewer proteins involved in ATP generation and oxidative phosphorylation. Basal metabolism in cultured primary ERRγ-negative cells ticked over as normal, but rose only about half as much as in wild-type neurons when the cells were stimulated with glucose. The result suggests ERRγ helps a neuron reach peak ATP production.
The researchers wondered if ERRγ is essential for learning and memory. Because ERRγ knockout mice die in just a few days, the researchers generated mice that selectively lost ERRγ in neurons of the cerebral cortex, hippocampus, and part of the olfactory bulb in adulthood. In these mice, long-term potentiation in the CA1 region of the hippocampus weakened considerably. Pyruvate, a breakdown product of glucose and starting material for oxidative phosphorylation, rescued this deficiency.
Behaviorally, the conditional ERRγ knockouts appeared normal, except for poor spatial memory. In the Morris water maze, they learned the location of a hidden platform more slowly than wild-type mice, and had trouble remembering where it was later on. Together the results suggest that reduced mitochondrial energy production is responsible for these deficits, the authors wrote.
Since mitochondrial defects in are linked to various diseases (Mattson et al., 2008), targeting ERRγ-dependent pathways with small molecules could offer new opportunities for therapy in various neurological disorders, the authors wrote. ERRγ has been classified as an orphan receptor, because its endogenous ligands are unknown, but researchers have developed small molecules that can stimulate it, such as GSK4716 (Zuercher et al., 2005). Others are attempting to make even more selective compounds (Kim et al., 2009). Pei plans to look at cell and animal models of Parkinson’s disease (PD), which has been tied to mitochondrial dysfunction, to see if knocking out ERRγ makes them more susceptible to illness (Jan 2007 news; Apr 2004 news). If it does, it might suggest that PD, and other diseases related to mitochondrial shortcomings, such as Alzheimer’s, might be treated with small molecules that specifically stimulate ERRγ function, he said.
Russell Swerdlow, University of Kansas Medical Center, Kansas City, agreed that the study has implications for drug development. He also pointed out that since ERRγ ignores genes responsible for breaking down fatty acids in the brain, it likely helps explain the brain’s dependence on glucose for energy rather than fatty acids, which are important for maintaining neuronal membranes. “Differences in these transcription factors probably explain a lot of this tissue-specific metabolism profiling.”—Gwyneth Dickey Zakaib
References
News Citations
- New Mouse Model Links Mitochondria to Parkinson Disease
- Pink Mutations Link Parkinson’s Disease to Mitochondria
Paper Citations
- Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009 Jan;89(1):121-45. PubMed.
- Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006 Jun;209(Pt 12):2304-11. PubMed.
- Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguère V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007 May;5(5):345-56. PubMed.
- Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008 Dec 10;60(5):748-66. PubMed.
- Zuercher WJ, Gaillard S, Orband-Miller LA, Chao EY, Shearer BG, Jones DG, Miller AB, Collins JL, McDonnell DP, Willson TM. Identification and structure-activity relationship of phenolic acyl hydrazones as selective agonists for the estrogen-related orphan nuclear receptors ERRbeta and ERRgamma. J Med Chem. 2005 May 5;48(9):3107-9. PubMed.
- Kim Y, Koh M, Kim DK, Choi HS, Park SB. Efficient discovery of selective small molecule agonists of estrogen-related receptor gamma using combinatorial approach. J Comb Chem. 2009 Sep-Oct;11(5):928-37. PubMed.
Further Reading
Papers
- Liu CC, Hu J, Tsai CW, Yue M, Melrose HL, Kanekiyo T, Bu G. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J Neurosci. 2015 Apr 8;35(14):5851-9. PubMed.
- Chen Z, Zhong C. Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog Neurobiol. 2013 Sep;108:21-43. PubMed.
- Wang Y, Wu L, Li J, Fang D, Zhong C, Chen JX, Yan SS. Synergistic exacerbation of mitochondrial and synaptic dysfunction and resultant learning and memory deficit in a mouse model of diabetic Alzheimer's disease. J Alzheimers Dis. 2015;43(2):451-63. PubMed.
- Franco-Iborra S, Vila M, Perier C. The Parkinson Disease Mitochondrial Hypothesis: Where Are We at?. Neuroscientist. 2015 Mar 11; PubMed.
Primary Papers
- Pei L, Mu Y, Leblanc M, Alaynick W, Barish GD, Pankratz M, Tseng TW, Kaufman S, Liddle C, Yu RT, Downes M, Pfaff SL, Auwerx J, Gage FH, Evans RM. Dependence of Hippocampal Function on ERRγ-Regulated Mitochondrial Metabolism. Cell Metab. 2015 Apr 7;21(4):628-36. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.