Does ApoE in Neurons Drive Selective Vulnerability in Alzheimer’s?
Quick Links
Scientists know that neurons can generate a dribble of ApoE, but it is considered a drop in the bucket relative to the massive amounts of it churned out by their glial neighbors. Nevertheless, a study published May 6 in Nature Neuroscience makes the case that neuronal ApoE not only signals distress, but also may have a hand in killing the neurons. Using single-cell transcriptomics, researchers led by Yadong Huang at the University of California, San Francisco, report that, in mice, certain neurons ratchet up ApoE expression as the animals age. They do so earlier, and with more gusto, in mice expressing ApoE4. Similarly, in people, particular neurons rev up ApoE in the early stages of Alzheimer’s disease. In later stages, those neurons were more likely to have perished, hinting that stepped-up ApoE expression could spell doom for neurons. Those same neurons also start expressing immune-response genes such as that for the antigen-presenting, major histocompatibility complex I. The researchers tied MHC-I to tau pathology within individual neurons.
- In mice, neurons that express ApoE also express antigen-presenting genes.
- This rises with age and, in ApoE4 knock-ins, starts earlier.
- Deleting ApoE from neurons wards off age-related death.
- In AD, neuronal ApoE tracks with progression, perhaps with cell loss.
“This fascinating paper describes a novel role for neuronal ApoE expression in Alzheimer’s disease,” wrote Gwyneth Welch and Li-Huei Tsai of the Massachusetts Institute of Technology (full comment below).
“The results leave little doubt that neurons can express substantial levels of ApoE transcripts in humanized mice and human postmortem brains, and that this expression is linked with pathology,” commented Tony Wyss-Coray of Stanford University in Palo Alto, California. “The study also opens a glimpse at what is likely to emerge from single-cell molecular profiling of brain cells in the years to come, and how it may finally provide answers to the perennial question of ‘selective cellular vulnerability.’”
Astrocytes make the lion’s share of ApoE in the brain, and in recent years, microglia as well have been found to ramp up ApoE expression when activated (Sep 2017 news). While most research labs have focused on glial sources of the apolipoprotein, Huang has studied its expression in neurons for many years. He initially used a GFP reporter mouse to show neurons boost ApoE expression in response to injury, and later linked neuronal ApoE to a slew of detrimental phenotypes, including Aβ and tau aggregation (Xu et al., 2006; timeline; Jun 2018 news). Huang also noticed that only some neurons, even within the same region of the brain, or of the same subtype, expressed ApoE.
Could those neurons be more susceptible to neurodegeneration? To test this idea, first author Kelly Zalocusky and colleagues asked how gene expression profiles defined different subsets of neurons in the hippocampus, and how those profiles changed with ApoE isoform and with age. They sequenced the nuclear transcriptomes of cells in ApoE3 knock-in and ApoE4-KI mice at 5, 10, 15, and 20 months of age. These animals carry human ApoE genes in place of their endogenous one. In all, the researchers measured transcripts of 21,204 genes from 123,489 mouse nuclei. Their gene expression profiles fell into 27 clusters of cells, including 16 distinct neuronal clusters. Lo and behold, more than any other transcript, expression of ApoE—regardless of ApoE genotype—best explained the variance among neuronal clusters. For example, using a principle component analysis, the researchers found that ApoE expression levels explained 73 percent of the variance among granule cells in the dentate gyrus.
Next, the researchers asked how ApoE expression relates to the rest of a neuron's transcriptome. Using pathway analysis, Zalocusky found that ApoE levels correlated with expression of genes involved in metabolism, neurodegeneration, cellular senescence, and apoptosis, all of which had previously been tied to neuronal ApoE expression. They also uncovered some new connections. For example, neuronal ApoE was linked to changes in DNA damage and repair, the unfolded protein response, and immune response genes. This last one came as a surprise, Huang said, because immune genes are typically considered the business of glial cells, not neurons.
To address how age affects ApoE expression in the context of transcriptome changes overall, the scientists calculated the proportion of neurons expressing the largest amount of ApoE within each of the 16 neuronal subtypes they had identified. They found that the proportion of these ApoE super-expressers rose with age, then fell again. What dictated this? Apparently, ApoE genotype. For example, in ApoE4-KI mice, the proportion of dentate gyrus granule cells and CA1 neurons that expressed abnormally high levels of ApoE peaked at 10 months of age, whereas for ApoE3-KI mice, the peak came at 15 months.
Is neuronal ApoE merely a marker of shifts in transcription, or does it drive damage and even death of the cell in which it happens? To find out, the researchers investigated how neuronal ApoE expression influenced age-related neuronal loss in ApoE-KI mice. They found that ApoE4-KI mice lost more neurons in the hippocampus with age than did ApoE3-KIs, and that the remaining neurons had fewer synapses. However, knocking out ApoE only from neurons prevented this age-related synaptic loss and neuronal death. Even ApoE3-KI mice benefited from getting of rid of neuronal ApoE, and their hippocampi better maintained their volume with age. This suggested that ApoE expression renders neurons vulnerable to degeneration.

No ApoE, Please. Older ApoE4-KI mice had fewer hippocampal synapses, as judged by PSD-95, than ApoE3-KIs. With ApoE removed from neurons (right two columns) the synapses stayed. [Courtesy of Zalocusky et al., Nature Neuroscience, 2021.]
Might immune response genes expressed in neurons replete with ApoE play a part in their vulnerability? Of all of the immune genes that tracked with ApoE, MHC-I genes correlated most strongly. The researchers attempted to pick apart the causal nature of these associations in primary neuron cultures, reporting that ApoE drove the uptick in MHC-I. Curiously, they also found that heightened ApoE expression made neurofibrillary tau appear in the neuronal soma. This was largely mediated by expression of functional MHC-I molecules on the cell surface.
Moreover, MHC-I appeared to exacerbate tau pathology in the mouse brain. The scientists triggered tau pathology in mice by injecting adeno-associated virus expressing human pathological tau into their hippocampi. Six weeks later, wild-type mice had more aggregated, phosphorylated human tau in the brains than did mice lacking β2-microglobulin, a protein required to stabilize MHC-I on the cell surface.
Does any of this hold up in the human brain? The researchers looked at single-nucleus transcriptomic data from 48 prefrontal cortex samples in the Religious Orders Study and Memory and Aging Project (ROSMAP). Astrocyte ApoE expression dwarfed that in neurons; still, some of the neurons did express more ApoE than their neighbors. The authors detected nine clusters of excitatory and four clusters of inhibitory neurons. The proportion of high ApoE expressors varied both by cluster and disease stage. At 28 percent, it was highest in one cluster of interneurons in people with mild cognitive impairment.
Jibing with the mouse data, the researchers reported that expression of genes in several biological pathways—most notably, immune response genes—correlated with ApoE transcripts in individual human neurons. They found the same associations in other datasets that included people without AD neuropathology. This suggested to them that, regardless of disease state, ApoE expression in a neuron was tied to that of immune genes. They did not find this relationship in non-neuronal cell types, including astrocytes.
In the ROSMAP cohort, which included people who had died at different stages of AD, neuronal ApoE expression tracked with disease stage. Among seven of the 13 neuronal subtypes, the proportion of cells that expressed much ApoE was highest among people with MCI, and lower in cognitively normal people and those with more advanced AD. Though cross-sectional, the findings look as if neuronal ApoE expression rises early in disease, then wanes in later stages.
Perhaps neurons expressing the most ApoE are those destined to die, and that’s why neuronal ApoE expression is lower with advanced disease? The ROSMAP data hint that this could be the case. Indeed, those neuronal subsets that had the highest proportion of ApoE super-expressors in people with MCI were sparser by the later stages of AD. In toto, the data suggest neurons might ramp up ApoE early in disease, and those that do are likely to succumb. Finally, in the ROSMAP part of this study, the researchers once again tied neuronal expression of several MHC-I genes with tau pathology.
“Altogether, these data indicate that the upregulation of ApoE–MHC-I pathways in individual neurons is a cellular response that eventually causes neuronal death, even during healthy ageing,” wrote Jessica Wagner and Jonas Neher of the German Center for Neurodegenerative Diseases in Tübingen in an editorial.
Huang and several commentators noted that the mechanisms connecting neuronal ApoE to MHC-I and to tangles are unclear; more study is needed to fill in the gaps. Many other cell pathways correlated with ApoE expression as well, so they could be involved, too. Earlier this year, researchers led by Martin Kampmann of the University of California, San Francisco, reported that excitatory neurons expressing the transcription factor RORB are particularly vulnerable to tau toxicity (Jan 2021 news).
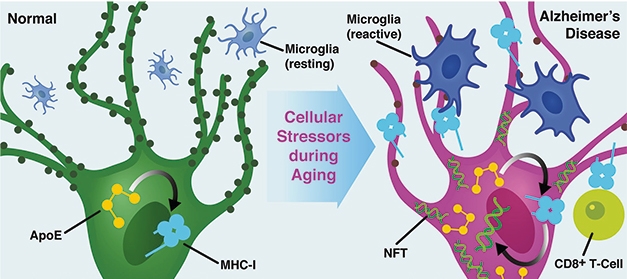
Stress to ApoE to MHC. In this proposed model of AD, normal neurons (green) increase expression of ApoE with age or stress (purple). This drives MHC-I expression, which, in turn, could trigger tau phosphorylation and attract CD8+ T cells or reactive microglia, which might prune synapses. [Courtesy of Zalocusky et al., Nature Neuroscience, 2021.]
“Selective vulnerability of specific neuronal subtypes is a hallmark of neurodegenerative diseases,” wrote Kampmann. “A systematic understanding of the underlying mechanisms would not only give us insights into disease mechanisms, but could also uncover therapeutic strategies to turn vulnerable neurons into resilient neurons.” His group plans to study the effect of ApoE genotype on RORB-linked vulnerability.
To Huang, the findings suggest that some neurons crank up ApoE under conditions of stress, including stress from aging and neuropathology. ApoE then somehow instigates a rise in immune response genes, including MHC-I, and this somehow instigates tau pathology—in neuronal culture. In the brain, a cadre of other players are involved. Huang hypothesized that MHC-I in neurons might serve as an “eat me” inviting immune cells to feast on synapses, reactivating a developmental pathway.
Michal Schwartz of the Weizmann Institute of Science in Tel-Aviv pointed out that neuronal expression of MHC-I has also been implicated in axonal regeneration following injury (Sabha et al., 2008; Joseph et al., 2011). “Expression of MHC-I by neurons is not all or none, good or bad, but rather, is dependent on context, timing, and persistence,” Schwartz wrote.
Conventionally speaking, MHC-I binds to T cell receptors on CD8+ T cells. Curiously, scientists led by Wyss-Coray recently spotted swarms of these cytotoxic cells in the brains of people with AD and PD (Jan 2020 news). “The studies open the possibility that ApoE may promote neuronal susceptibility to potential T cell killing by upregulating the cognate receptor (i.e., MHC-I-b2M-peptide) for virus-specific T cells, or T cells of yet unknown specificity for neuronal antigens,” Wyss-Coray noted.
If neuronal stress is what triggers ApoE expression, then whence the stress, and why does it happen sooner in ApoE4-expressing mice? Guojun Bu of the Mayo Clinic in Jacksonville, Florida, said aging brings on myriad stresses in the brain. They run the gamut from oxidative to erosion of the vasculature to sluggish clearance of harmful metabolites. ApoE4 may render the brain more vulnerable to these age-related stressors, he said.
Sam Gandy and Michelle Ehrlich of Mount Sinai School of Medicine in New York wondered how to square Zalocusky’s results on neuronal ApoE4 with those by David Holtzman’s group, which reported that selectively removing ApoE4 from astrocytes protected neurons in a mouse model of tauopathy (Apr 2021 news). No matter where it is expressed, ApoE4 seems to harm the brain. “As a result, one wonders whether whole-body knockdown of APOE expression might be the most parsimonious solution to what has turned out to be a huge challenge of ‘APOE-e4-whack-a-mole,’” Gandy and Ehrlich wrote.
To Ole Isacson of Harvard Medical School, the data suggest that neurons are compensating for the shortcomings of their glial support staff. Among a plethora of functions, astrocytes normally supply neurons with lipids, delivered in an ApoE package, which neurons use to replenish their extensive membranes. “If coupling between astrocyte and neuron in terms of lipid handling is faulty, the neuron is going to go under,” Isacson said. Perhaps neurons cope by churning out their own ApoE. In this context, Isacson said, neuronal ApoE might benefit the neuron, at least for awhile. Because ApoE4 is worse at carrying lipids than is ApoE3, neurons in ApoE4-KI mice may start to compensate earlier.
Isacson emphasized that beyond lipid handling, ApoE is also known to promote the resolution of inflammation, through binding to the complement protein C1q (Feb 2019 news). He therefore cautioned against the idea of targeting the apolipoprotein as a therapeutic strategy.—Jessica Shugart
References
News Citations
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- In Human Neurons, ApoE4 Promotes Aβ Production and Tau Phosphorylation
- Selective Vulnerability News: RORB Neurons Are First Victims of Tangles
- Attack of the Clones? Memory CD8+ T Cells Stalk the AD, PD Brain
- Squelching ApoE in Astrocytes of Tau-Ravaged Mice Dampens Degeneration
- ApoE Binds Complement Protein, Keeps Inflammatory Cascade in Check
Paper Citations
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006 May 10;26(19):4985-94. PubMed.
- Sabha M Jr, Emirandetti A, Cullheim S, De Oliveira AL. MHC I expression and synaptic plasticity in different mice strains after axotomy. Synapse. 2008 Feb;62(2):137-48. PubMed.
- Joseph MS, Bilousova T, Zdunowski S, Wu ZP, Middleton B, Boudzinskaia M, Wong B, Ali N, Zhong H, Yong J, Washburn L, Escande-Beillard N, Dang H, Edgerton VR, Tillakaratne NJ, Kaufman DL. Transgenic mice with enhanced neuronal major histocompatibility complex class I expression recover locomotor function better after spinal cord injury. J Neurosci Res. 2011 Mar;89(3):365-72. Epub 2010 Dec 22 PubMed.
Other Citations
Further Reading
No Available Further Reading
Primary Papers
- Zalocusky KA, Najm R, Taubes AL, Hao Y, Yoon SY, Koutsodendris N, Nelson MR, Rao A, Bennett DA, Bant J, Amornkul DJ, Xu Q, An A, Cisne-Thomson O, Huang Y. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer's disease. Nat Neurosci. 2021 Jun;24(6):786-798. Epub 2021 May 6 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Picower Institute for Learning and Memory
Picower Institute of MIT
This fascinating paper from the Huang lab describes a novel role for neuronal ApoE expression in Alzheimer’s disease.
Zalocusky and colleagues find that neuronal ApoE is linked to immune response activation and selective neurodegeneration. Specifically, they show that increased ApoE drives the expression of MHC-I, which leads to tau pathology as well as synapse and neuronal loss. These compelling data provide mechanistic insight to cell-type vulnerability in Alzheimer’s disease. Because the relationship between neuronal ApoE and immune gene expression is also found in individuals without pathology or neurodegeneration, it will be useful to define how ApoE interacts with disease-associated biological pathways to drive degeneration phenotypes. It will also be interesting to see how microglia and other glial cells respond to aberrant MHC-I expression.…More
Overall, this is an important and thought-provoking paper that raises numerous questions regarding the mechanisms of cell type vulnerability and neuroinflammation.
Weizmann Institute of Science
Selectivity of neuronal loss under disease conditions is still a mystery. In the present study, Zalocusky and colleagues attribute a key role to neuronal expression of APOE in regulating MHC-I expression, and thereby, selective neuronal vulnerability. This new concept regarding neuronal vulnerability is interesting in light of the emerging understanding of the role displayed by the immune system in supporting life-long brain plasticity and repair (Moalem et al., 1999; Rapalino et al., 1998; Ziv et al., 2006; Baruch et al., 2015; Filiano et al., 2016), including its effects in neurodegenerative diseases.
Neuronal expression of APOE (Wang et al., 2018; Xu et al., 1999) and MHC-I (Lee et al., 2014) are new observations and are still far from being fully understood. Specifically, ApoE4 expression has been identified as a risk factor in human patients suffering from AD; the severity of the disease is greater in patients carrying APOE4 allele (Tao et al., 2018). In a mouse model of tauopathy, human ApoE expression—especially of the ApoE4—led to increased tau pathology, neuroinflammation, and neuronal loss.…More
Regarding MHC-I, it has generally been presumed that it is not expressed by neurons under physiological conditions (Cebrián et al., 2014). The initial study that showed MHC-I gene expression by neurons in response to IFN-γ was reported in 1995 (Neumann et al., 1995). Other studies suggested that neuronal expression of this molecule is involved in neuroinflammatory processes, and participates in immune-mediated neurodegeneration. However, accumulating data have subsequently demonstrated MHC-I expression by subsets of neurons in both adult and developing mammalian brains, even under physiological conditions (Lindå et al., 1999; Huh et al., 2000; Letellier et al., 2008). Neuronal MHC-I expression was further implicated in models of axonal regeneration, where it was shown to regulate the ability of neurons to regenerate axons (Sabha et al., 2008). Moreover, it was suggested that neuronal expression of MHC-I during the first week after sterile injury enhances axonal regeneration, and these findings support the observation that elevated neuronal MHC-I expression promotes the recovery of locomotor abilities after spinal cord injury (Joseph et al., 2011). In light of these results, it is clear that expression of MHC-I by neurons it is not all or none, good or bad, but rather, is dependent on context, timing, and persistence.
The proposed mechanism in the present study, that neuronal ApoE expression might be an important factor driving within-neuron-type variability under both normal physiological and pathophysiological conditions, is intriguing. The authors further showed that reducing functional MHC-I by knocking down or knocking out B2M, a protein required for functional expression of all MHC-I genes, is sufficient to significantly reduce tau pathologies in vitro in cultured primary neurons, or in vivo in a pathological tau-P301S overexpression mouse model.
In this regard, it is interesting to note that B2M expression was found to be dependent on Type I interferon (Baruch et al., 2014; Deczkowska et al., 2017), the level of which is chronically elevated in aging and neurodegenerative conditions; it is therefore quite possible that MHC-I expression is regulated by Type I inflammation. While the functional connection between APOE and MHC-I expression by neurons is interesting, further studies are needed to fully dissect out the causal relationships connecting APOE expression with adaptive immunity, and the implications to all forms of APOE.
References:
Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999 Jan;5(1):49-55. PubMed.
Rapalino O, Lazarov-Spiegler O, Agranov E, Velan GJ, Yoles E, Fraidakis M, Solomon A, Gepstein R, Katz A, Belkin M, Hadani M, Schwartz M. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med. 1998 Jul;4(7):814-21. PubMed.
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006 Feb;9(2):268-75. PubMed.
Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, Tsitsou-Kampeli A, Sarel A, Cahalon L, Schwartz M. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun. 2015 Aug 18;6:7967. PubMed.
Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, Overall CC, Gadani SP, Turner SD, Weng Z, Peerzade SN, Chen H, Lee KS, Scott MM, Beenhakker MP, Litvak V, Kipnis J. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature. 2016 Jul 21;535(7612):425-9. Epub 2016 Jul 13 PubMed.
Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, Yoon SY, Yuan H, Li G, Miller ZA, Miller BL, Malloy MJ, Huang Y. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018 May;24(5):647-657. Epub 2018 Apr 9 PubMed.
Xu PT, Gilbert JR, Qiu HL, Ervin J, Rothrock-Christian TR, Hulette C, Schmechel DE. Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol. 1999 Feb;154(2):601-11. PubMed.
Lee H, Brott BK, Kirkby LA, Adelson JD, Cheng S, Feller MB, Datwani A, Shatz CJ. Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature. 2014 May 8;509(7499):195-200. Epub 2014 Mar 30 PubMed.
Tao Q, Ang TF, DeCarli C, Auerbach SH, Devine S, Stein TD, Zhang X, Massaro J, Au R, Qiu WQ. Association of Chronic Low-grade Inflammation With Risk of Alzheimer Disease in ApoE4 Carriers. JAMA Netw Open. 2018 Oct 5;1(6):e183597. PubMed.
Cebrián C, Loike JD, Sulzer D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson's and other brain diseases. Front Neuroanat. 2014;8:114. Epub 2014 Oct 13 PubMed.
Neumann H, Cavalié A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science. 1995 Jul 28;269(5223):549-52. PubMed.
Lindå H, Hammarberg H, Piehl F, Khademi M, Olsson T. Expression of MHC class I heavy chain and beta2-microglobulin in rat brainstem motoneurons and nigral dopaminergic neurons. J Neuroimmunol. 1999 Nov 1;101(1):76-86. PubMed.
Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000 Dec 15;290(5499):2155-9. PubMed.
Letellier M, Willson ML, Gautheron V, Mariani J, Lohof AM. Normal adult climbing fiber monoinnervation of cerebellar Purkinje cells in mice lacking MHC class I molecules. Dev Neurobiol. 2008 Jul;68(8):997-1006. PubMed.
Sabha M Jr, Emirandetti A, Cullheim S, De Oliveira AL. MHC I expression and synaptic plasticity in different mice strains after axotomy. Synapse. 2008 Feb;62(2):137-48. PubMed.
Joseph MS, Bilousova T, Zdunowski S, Wu ZP, Middleton B, Boudzinskaia M, Wong B, Ali N, Zhong H, Yong J, Washburn L, Escande-Beillard N, Dang H, Edgerton VR, Tillakaratne NJ, Kaufman DL. Transgenic mice with enhanced neuronal major histocompatibility complex class I expression recover locomotor function better after spinal cord injury. J Neurosci Res. 2011 Mar;89(3):365-72. Epub 2010 Dec 22 PubMed.
Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, Amit I, Schwartz M. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014 Aug 21; PubMed.
Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, Singer O, David E, Winter DR, Smith LK, Kertser A, Baruch K, Rosenzweig N, Terem A, Prinz M, Villeda S, Citri A, Amit I, Schwartz M. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun. 2017 Sep 28;8(1):717. PubMed.
Stanford University Medical School
This is another pearl from the Yadong Huang lab at the Gladstone Institutes/UCSF cementing the hypothesis that neurons express ApoE, that this expression is genotype-dependent (APOE4 more expressed than APOE3), and that it is detrimental and linked to pathology. Most in the field initially dismissed a (patho)physiological role for neuronal ApoE in neurodegeneration, but the Huang lab provided ever-increasing experimental evidence employing, in this current study, single cell/nucleus RNA sequencing of individual neurons. The results leave little doubt that neurons can express substantial levels of APOE transcripts in humanized mice and human postmortem brains and that this expression is linked with pathology. The study also opens a glimpse in what is likely to emerge from single-cell molecular profiling of brain cells in the years to come, and how it may finally provide answers to the perennial question of “selective cellular vulnerability.”…More
Intriguingly, ApoE expression at the single cell/nucleus level strongly correlates with expression of immune pathway genes, most notably MHC class I. Genetic deletion of human APOE in neurons of APOE humanized mice protects them from ApoE4 induced neurodegeneration and lowered MHC-I levels as well as related genes including Tap2, which is involved in peptide loading on the MHC-I/b2-microglobulin (B2M) complex.
Zalocusky and colleagues further show that ApoE expression correlates with tau pathology—an observation they made earlier—and that this is MHC-I dependent. Because B2M is necessary for stable cell surface expression of MHC-I they were able to lower expression of MHC-I by genetically reducing B2M expression in neurons and show that this too, leads to reduced tau pathology in cell culture and in mice. Together, the authors propose a cascade whereby APOE4 and/or stress increase neuronal ApoE expression, which then leads to increased MHC-I surface expression and subsequent abnormal tau phosphorylation and neurodegeneration.
This exciting new pathway offers potential new therapeutic targets for neurodegenerative diseases. It is interesting that B2M rises prominently with normal aging, impairs neurogenesis, and promotes cognitive impairment in young mice, while B2M deletion improves cognition in aged mice (Smith et al., 2015). Our recent observation that virus-specific, CD8-positive, T effector cells are clonally expanded in the cerebrospinal fluid of Alzheimer's disease patients, and can be localized to degenerating neurons in brain parenchyma, opens the possibility that ApoE may promote neuronal susceptibility to potential T cell killing by upregulating the cognate receptor (i.e., MHC-I-B2M-peptide) for virus-specific T cells or T cells of yet unknown specificity for neuronal antigens (Gate et al., 2020).
ApoE-driven MHC-I expression, as the authors discussed, could also interfere with synaptic plasticity as laid out in the elegant cascade uncovered by Carla Shatz and colleagues at Harvard Medical School (Huh et al., 2000). Future studies need to disentangle these possibilities and assign pathological relevance to them; equally important, we need to understand how ApoE regulates MHC-I expression and how this promotes tau phosphorylation at a mechanistic molecular level.
References:
Smith LK, He Y, Park JS, Bieri G, Snethlage CE, Lin K, Gontier G, Wabl R, Plambeck KE, Udeochu J, Wheatley EG, Bouchard J, Eggel A, Narasimha R, Grant JL, Luo J, Wyss-Coray T, Villeda SA. β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat Med. 2015 Aug;21(8):932-7. Epub 2015 Jul 6 PubMed.
Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, Chen K, Lehallier B, Channappa D, De Los Santos MB, McBride A, Pluvinage J, Elahi F, Tam GK, Kim Y, Greicius M, Wagner AD, Aigner L, Galasko DR, Davis MM, Wyss-Coray T. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer's disease. Nature. 2020 Jan;577(7790):399-404. Epub 2020 Jan 8 PubMed.
Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000 Dec 15;290(5499):2155-9. PubMed.
Harvard Medical School
An important finding in this study is the demonstration of clear correlations between neuronal ApoE expression and neuronal phenotype in humanized ApoE-KI mouse models, involving shifts in cellular metabolism, immune response, and pathways linked to neurodegenerative diseases. Neuronal pathways involved in cellular metabolism and immune response also correlated with ApoE expression in human patients with MCI or AD. Another key observation is that in humanized ApoE-KI mice expressing ApoE3 or ApoE4, the subpopulation of high-ApoE-expressing neurons increases with age at first, and then sharply declines. Interestingly, they show that both the increase and the decline occur sooner in ApoE4-KI mice compared to ApoE3-KI mice. In addition, results show a correlation in neurons between ApoE and MHC-I expression, suggestive of neuroimmune regulatory pathway interactions.…More
While the cellular gene-signature approaches used as a basis for the authors’ explorations are very useful, there are several weaknesses in the representation of the data and the conceptual over-simplifications that follow. The conclusion that ApoE expression increases in neurons that are pathological may be too simplistic, since the increases more likely reflect a change from physiological control of lipid transfer from astrocytes to neurons—a pathophysiological adaptation to the failure of glial coping mechanisms against other causes of neuronal stress. High-ApoE-expressing neurons do not mean ApoE causes vulnerability.
The authors describe populations of high-ApoE-expressing neurons in MCI and AD patients and interpret this as evidence that increased ApoE expression causes selective vulnerability. While these high-ApoE-expressing neurons may be selectively vulnerable, they do not consider that increased ApoE expression may itself be a stress response, rather than pathological.
Conceptually critical, there is no evidence presented in the data that the known aging and AD regional brain and individual neuronal vulnerability (relative vulnerability) differences are reflected in the measured ApoE expression profiles. Instead, the authors treat all “neurons” as equal in the brain. Nuclear neuronal gene signatures are all lumped together, while we know that MCI and AD have regional, cellular, and staged pathological progression that depends on the individual neuronal cell types. Such neuronal vulnerability is not necessarily “selective,” but relative. Not all brain neurons degenerate in aging, MCI, and AD. Brain neurons are heterogenous and can be categorized by neurotransmitter, anatomy, and gene-expression profiles. The relative neuronal cell type degeneration risks vary for each neuronal cell type and brain region depending on the underlying and real vulnerability to neuronal aging and AD risk factors. Therefore, the data shown, that neurons in general increase (no regional differences/clustering were observed) their ApoE expression may simply reflect initial neuronal adaptive responses to the underlying neuronal and glial causes of pathology, independent of ApoE levels.
The association found in the report between ApoE levels and MHC-I is interesting due to the well-known role of ApoE in resolution of inflammatory processes and metabolic control under stress. Numerous previous studies have demonstrated that ApoE can cross-talk with components of the immune system, including C1q. These prior relevant pathophysiological results are not discussed or referenced. For example, Yin et al demonstrated that ApoE can functionally attenuate C1q activation under physiological lipid stress in vivo (Yin et al., 2019). Also not mentioned are other important studies (by Maria Ioannou in particular) showing that there is a necessary functional cross-talk between astrocytes and neurons in lipid transfer, where the astrocytes play the normal key physiological role (Ioannou et al., 2019; Qi et al., 2021; Sienski et al., 2021). Given that the authors themselves show that the majority of ApoE expression (~ fivefold higher) occurs in astrocytes/glia cell clusters, also with time, the changes observed in neurons are likely the tip of the iceberg in terms of the actual ApoE lipid and lipid transfer in a pathophysiological context.
In conclusion, while this article has numerous interesting observations, key questions remain about the pathophysiological roles of the lipid binding protein ApoE and its human allele forms ApoE 2, 3, and 4, and how to mitigate the inherent risk of each ApoE form or their loss of function.
References:
Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, Pasolli HA, Xu CS, Pang S, Matthies D, Hess HF, Lippincott-Schwartz J, Liu Z. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell. 2019 May 30;177(6):1522-1535.e14. Epub 2019 May 23 PubMed.
Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021 Jan 5;34(1):108572. PubMed.
Sienski G, Narayan P, Bonner JM, Kory N, Boland S, Arczewska AA, Ralvenius WT, Akay L, Lockshin E, He L, Milo B, Graziosi A, Baru V, Lewis CA, Kellis M, Sabatini DM, Tsai LH, Lindquist S. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med. 2021 Mar 3;13(583) PubMed.
Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, Nietzsche S, Westermann M, Peng L, Hu D, Bontha SV, Srikakulapu P, Beer M, Megens RT, Steffens S, Hildner M, Halder LD, Eckstein HH, Pelisek J, Herms J, Roeber S, Arzberger T, Borodovsky A, Habenicht L, Binder CJ, Weber C, Zipfel PF, Skerka C, Habenicht AJ. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019 Mar;25(3):496-506. Epub 2019 Jan 28 PubMed.
Make a Comment
To make a comment you must login or register.