ApoE Binds Complement Protein, Keeps Inflammatory Cascade in Check
Quick Links
This story was updated on February 5, 2019.
Just when you thought ApoE biology couldn’t get any more complicated, scientists have discovered a new function for this apolipoprotein. According to a January 28 paper in Nature Medicine, ApoE keeps the classical complement cascade in check by binding to a key activator, C1q. Researchers led by Changjun Yin and Andreas Habenicht of Ludwig-Maximilians-University in Munich and Christine Skerka of the Leibniz Institute in Jena, Germany, also spotted the ApoE-C1q complexes in three places: commingling with lipid deposits in both the choroid plexus and atherosclerotic arteries, and with Aβ plaques in the cortex. In ApoE-knockout mice, complement activation ran rampant. Suppressing complement protein C5 curbed the infiltration of inflammatory cells in these regions in animal models.
- ApoE binds to C1q, blocking initiation of the classical complement cascade.
- ApoE-C1q complexes are around lipid deposits in the choroid plexus, near Aβ plaques, and in arterial plaques.
- Shutting down complement protein C5 ameliorated harmful inflammation.
“The authors demonstrate that ApoE’s high-affinity binding to the C1q protein inhibits the classical complement cascade, a clear example of a disease-modifying effect of ApoE not directly related to amyloid,” commented Bill Rebeck of Georgetown University in Washington, D.C. Yadong Huang of the University of California, San Francisco, noted that the study links two prominent risk factors for AD: ApoE and inflammation.
“They present convincing evidence that ApoE is a checkpoint inhibitor for complement activation once complement activation is underway,” commented Cynthia Lemere of Brigham and Women’s Hospital in Boston. “This seems to be a common pathway for both AD and atherosclerosis.”
The best-known function of ApoE is that of lipid transporter: It sops up circulating lipids and facilitates their uptake and metabolism by cells. The E4 isoform boosts AD risk, and researchers have proposed myriad mechanisms to explain this, including that ApoE4 hinders Aβ clearance while ramping up its production (Apr 2013 news; Jun 2018 news; Jan 2017 news). Studies have also implicated E4 in messing with synaptic pruning, and exacerbating neuroinflammatory responses against tau pathology (Sep 2016 news; Apr 2017 news). ApoE4 carriers also have higher levels of plasma cholesterol, putting them at greater risk for atherosclerosis.
The German researchers hypothesized that ApoE might figure in the pathogenesis of AD and atherosclerosis by a common mechanism. To investigate, they started by examining ApoE in the choroid plexus. The ChP forms a barrier between the blood and cerebrospinal fluid (CSF), and is one of the key ports of entry for immune cells recruited to the brain. Previous studies indicated that ApoE expression is high in the ChP, and that increased inflammation there causes cognitive deficits with age (Aug 2014 news). Co-first authors Yin, Habenicht, and colleagues found that in ApoE knockout mice—which have sky-high levels of circulating lipids—the ChP was chock-full of lipid deposits, and massively infiltrated by leukocytes. Skerka thinks the blood-CSF barrier was compromised as well, because the researchers detected immunoglobulins in the ChP.
Amidst this greasy mess, the scientists also spotted abundant complement proteins, including C3, C3a, and C5. C5 is primarily produced in the liver. It is required to amplify recruitment of immune cells to the site of complement activation, and to form the attack complex that ultimately punctures target cells, such as microbes. When the researchers turned down C5 expression in the liver, dramatically fewer leukocytes reached the ChP in the ApoE knockouts. In all, the findings suggested that without ApoE, lipid build-up in the ChP tripped off the complement cascade there, leading to infiltration of immune cells.

Lipids in Choroid Plexus. Lipid deposits (red) accumulated in the choroid plexus of ApoE knockouts, and in ApoE4 mice on a high-fat diet (HFD). [Courtesy of Yin et al., 2019.]
Would a similar scenario unfold in the presence of human ApoE isoforms? To find out, the researchers looked at the ChPs of mice expressing human ApoE3 or ApoE4 instead of mouse ApoE. Similar to wild-type, neither of these mouse models had lipid deposits, complement proteins, nor leukocyte infiltration in the ChP. However, this changed when the researchers put the ApoE4 mice on a high-fat diet. With plasma cholesterol levels rivaling those of ApoE knockouts, these mice developed abundant lipid deposits in their ChPs. However, they had only minimal activation of the complement cascade. This suggested that though lipids built up in the ChPs of ApoE4 mice eating fatty chow, the presence of ApoE4 still kept the complement cascade in check there.
How so? Co-first author Susanne Ackermann and colleagues from the Skerka lab investigated this question in vitro. They exposed typical complement targets, including E. Coli, to normal human serum, which is naturally steeped in complement proteins. All three isoforms of ApoE effectively spared these targets from lysis. Oxidized low-density lipoproteins and Aβ fibrils also kicked off the complement cascade in human serum, but ApoE isoforms blocked that, too. Using a slew of structural and biochemical techniques, the researchers found that ApoE bound to C1q. A peptide containing residues 139-152 of ApoE bound with high affinity to the stalk region of the activated form of C1q only: C1q exists in an inactive form until encountering a target, then changes conformation. Notably, all three isoforms of ApoE bound to C1q with equal affinity.
How does all this relate to the different effects of ApoE isoforms on AD risk, then? “That’s the key unresolved question here,” said Huang, who was intrigued that all ApoE isoforms appeared to bind and inhibit C1q equally. Skerka proposed that the isoforms modulate AD risk by differentially influencing complement activators, such as lipids and Aβ. Even if all isoforms inhibit the cascade with equal vigor, perhaps accumulating triggers eventually overwhelm this checkpoint in ApoE4 carriers on a high fat diet, for example, she suggested.

Lipid Nexus in the Plexus? The choroid plexus in people with dementia (right) had more lipid deposits (red) than did the ChP of normal controls (left). [Courtesy of Yin et al., 2019.]
The researchers next looked for evidence of the ApoE-C1q connection in human postmortem brain tissue from Neurobiobank Munich. The samples came from 30 people, averaging 70 years of age at death, 10 of whom carried at least one copy of ApoE4. Seventeen had been diagnosed with dementia, and also had significant burdens of Aβ and tau pathology, while 13 had not been diagnosed with dementia and had little to no evidence of these pathologies. The researchers found lipid deposits in 29 out of 30 ChP samples. The burden of ChP lipid deposits was highest in people with dementia, and correlated with the extent of Aβ and tau pathology. On average, ApoE4 carriers, all but one of whom had dementia, had the highest ChP lipid load. Using proximity labeling, they found ApoE-C1q complexes mingling within the lipid deposits.
The researchers also detected numerous complement proteins as well as ApoE around plaques, as reported previously. Using proximity labeling, they found that ApoE formed complexes with C1q there, as well. C1q has been reported to nucleate Aβ aggregation (Webster et al., 1995). Yin also found ApoE-C1q complexes in and around plaques in APP/PS1 mice. In those animals, knocking down liver expression of C5 reduced the number of microglia surrounding plaques, suggesting that microglial recruitment to plaques is at least partly driven by the complement cascade and that ApoE cannot completely shut it down. Lemere noted that this finding agrees with her studies in C3-deficient APP/PS1 mice, which also had fewer microglia surrounding plaques. Shutting down the cascade did not affect the overall plaque burden.
What about in atherosclerosis? ApoE knockout mice eventually develop atherosclerosis, and the researchers compared gene expression profiles in the early stages of the disease to wild-type mice. They found numerous complement proteins upregulated in the atherosclerotic arteries. Knocking down C5 expression in these mice reduced atherosclerosis by 65 percent, suggesting the complement cascade played a major role in the disease.
The researchers examined postmortem carotid artery samples from five healthy control, six early, and nine advanced atherosclerotic arteries. They found activated macrophages along with ApoE, C1q, and C5 within atherosclerotic plaques in the early and advanced stages. Notably, while both ApoE and C1q were expressed in the uninflamed areas of the arterial wall, they only formed complexes around plaques.
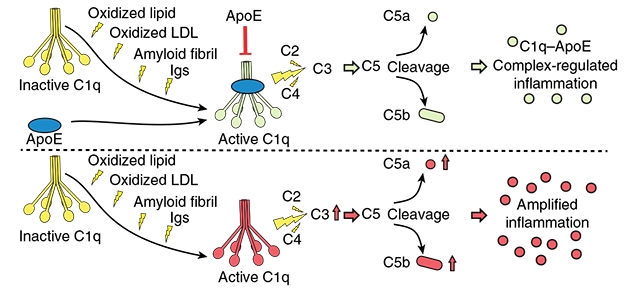
ApoE Checks C1q. In the proposed model, multiple triggers, including oxidized lipids and Aβ, activate C1q, initiating the complement cascade. ApoE binds and inhibits C1q. [Courtesy of Yin et al., 2019.]
Habenicht believes the complement cascade could be a common therapeutic target in both AD and atherosclerosis. How ChP lipid deposits relate to AD pathology remains unclear. Larger human studies need to be done, Habenicht added.
Lemere wondered whether an overactive complement cascade could explain amyloid-related imaging abnormalities (ARIA) that occur in some people in response to Aβ immunotherapy, especially in ApoE4 carriers. “Perhaps ApoE4 carriers have diminished ability for ApoE to act as a complement checkpoint inhibitor,” she wrote. “If so, reducing complement activation … might be an effective way to combat ARIA.”
To Huang’s mind, that the relationship between ApoE and inflammation, and the role of inflammation in neurodegeneration, is not clear-cut. For example, recent studies suggest that ApoE, and ApoE4 more so, exacerbates harmful inflammatory responses against tau, ramping up neurodegeneration in tauopathy models (Sep 2017 news). How does this square with the present data, which cast ApoE as a soother of inflammation? The findings open up a new area for investigation, he said.—Jessica Shugart
References
News Citations
- ApoE Does Not Bind Aβ, Competes for Clearance
- In Human Neurons, ApoE4 Promotes Aβ Production and Tau Phosphorylation
- ApoE Risk Explained? Isoform-Dependent Boost in APP Expression Uncovered
- ApoE Variants Modulate Astrocyte Appetite for Synapses
- ApoE and Tau: Unholy Alliance Spawns Neurodegeneration
- Choroid Plexus May Hold a Key To Aging Brain
- ApoE4 Makes All Things Tau Worse, From Beginning to End
Research Models Citations
Paper Citations
- Webster S, Glabe C, Rogers J. Multivalent binding of complement protein C1Q to the amyloid beta-peptide (A beta) promotes the nucleation phase of A beta aggregation. Biochem Biophys Res Commun. 1995 Dec 26;217(3):869-75. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, Nietzsche S, Westermann M, Peng L, Hu D, Bontha SV, Srikakulapu P, Beer M, Megens RT, Steffens S, Hildner M, Halder LD, Eckstein HH, Pelisek J, Herms J, Roeber S, Arzberger T, Borodovsky A, Habenicht L, Binder CJ, Weber C, Zipfel PF, Skerka C, Habenicht AJ. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019 Mar;25(3):496-506. Epub 2019 Jan 28 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Georgetown University
The work identifies a mechanism for the known anti-inflammatory effects of ApoE through a series of approaches. The authors demonstrate that ApoE’s high-affinity binding to the C1q protein inhibits the classical complement cascade, a clear example of a disease-modifying effect of ApoE not directly related to amyloid. The work also shows interesting early pathological changes in the choroid plexus, with an accumulation of lipids and leukocytes at this blood-CSF barrier. Interestingly, these effects are precipitated by a combination of the strong genetic (ApoE4) and environmental (high-fat diet) risk factors for Alzheimer’s disease. The therapeutic usefulness of these findings is demonstrated in the binding of an ApoE-derived peptide to C1q.…More
This mechanistic insight fits with previous research indicating ApoE peptides decrease inflammation and provide neuroprotection across several neuropathological conditions. Together the data support a model of loss of ApoE function in the ApoE4 isoform and suggest that research into the detrimental effects of ApoE (in the presence of environmental stresses) is a useful approach to AD-prevention strategies.
Ann Romney Center for Neurologic Diseases, Brigham and Women's Hospital and Harvard Medical School
This is quite a paper! The authors present convincing evidence that ApoE is a checkpoint inhibitor for complement activation once complement activation is underway. This seems to be a common pathway for both AD and atherosclerosis. This is very interesting, as vascular disease is highly prominent in Alzheimer’s disease. The paper is thorough and intriguing, and really got the wheels turning in my brain.
Their data with the C5 siRNA treatment in APP PS1 21 mice is quite similar to our data in germline-C3-deficient APP/PS1dE9 mice, in which microglia association with plaques was significantly reduced at 16 months of age. It would be interesting to go back to those mouse brains and look for Aβ/ApoE and C1q/ApoE complexes near or within plaques.…More
It would also be interesting to look at brain tissue from people who developed ARIA following Aβ immunotherapy. Antibody-antigen complexes activate the classical complement cascade. Perhaps ApoE4 carriers have diminished ability for ApoE to act as a complement checkpoint inhibitor. This might result in exacerbated chronic inflammation which might then lead to vasogenic edema and/or microhemorrhage following early boluses of antibody, especially in people with the ApoE4 genotype. If so, this would suggest that reducing complement activation—either by modification of antibodies or something like siRNA targeting complement—might be an effective way to combat ARIA side effects in immunotherapy. An interesting question would be whether C1q/ApoE complexes are associated with vascular amyloid in AD ApoE4 versus non-E4 carriers.
It would be interesting to know whether there are sex differences in the ability of ApoE to inhibit complement. Female ApoE4 carriers seem to be at higher risk of AD, have more Aβ, and progress more quickly than males with the same ApoE4 genotype.
Finally, the presence of ApoE/Aβ complexes but lack of ApoE/p-tau complexes in plaques in AD brain seems consistent with a report by Bonham et al. (2016) in which they found a significant interaction between CSF ApoE4 and C3 on amyloid and p-tau, but C3 was only associated with p-tau after accounting for Aβ (in CSF). Notably, there is evidence that C1q can bind directly to Aβ (Webster et al., 1995)!
References:
Bonham LW, Desikan RS, Yokoyama JS, Alzheimer’s Disease Neuroimaging Initiative. The relationship between complement factor C3, APOE ε4, amyloid and tau in Alzheimer's disease. Acta Neuropathol Commun. 2016 Jun 29;4(1):65. PubMed.
Webster S, Glabe C, Rogers J. Multivalent binding of complement protein C1Q to the amyloid beta-peptide (A beta) promotes the nucleation phase of A beta aggregation. Biochem Biophys Res Commun. 1995 Dec 26;217(3):869-75. PubMed.
Weizmann Institute of Science
The present study highlights the connection between cholesterol, the choroid plexus and immune-brain control communication. Yin et al. attribute a key role to APOE in controlling the choroid plexus (ChP), as a functional interface between the brain and the circulating immune cells. Specifically, the authors demonstrate that ApoE acts as the rheostat regulating the classical complement cascade (CCC) following direct binding to C1q, which according to the authors controls leukocyte infiltration into the brain and inflammation. An important implication is the relevance of lipid homeostasis as a marker for the ChP gateway activity. As an apolipoprotein, the primary role of ApoE is lipid binding and transport. Notably, Yin et al. connected locally impaired lipid homeostasis at the ChP to neuroinflammation. Remarkably, a similar phenotype was observed in mice carrying the AD-associated allele ApoE4 when subjected to a high-fat diet (HFD), but not in mice carrying the ApoE3 allele.…More
In light of the new data that were recently generated in connection with the ChP, one of the important findings that emerges from the present study is the observation that ChP shows a Type I interferon signature under conditions that impose APOE dysfunctions. In particular, the balance between type I and type II interferon was found to be crucial for the correct functioning of the ChP, with type I interferon responses characterizing altered physiological states such as aging (Baruch et al., 2014; Deczkowska et al., 2017) and Alzheimer’s disease (Baruch et al., 2015; Mesquita et al., 2015). In light of this, one might suggest that Type I interferons might be upregulated in AD patients carrying the APOE4 allele, which could explain their more severe disease.
Stefano Suzzi and Afroditi Tsitsou-Kampeli are co-authors of this comment
References:
Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, Amit I, Schwartz M. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014 Aug 21; PubMed.
Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, Singer O, David E, Winter DR, Smith LK, Kertser A, Baruch K, Rosenzweig N, Terem A, Prinz M, Villeda S, Citri A, Amit I, Schwartz M. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun. 2017 Sep 28;8(1):717. PubMed.
Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, Tsitsou-Kampeli A, Sarel A, Cahalon L, Schwartz M. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun. 2015 Aug 18;6:7967. PubMed.
Mesquita SD, Ferreira AC, Gao F, Coppola G, Geschwind DH, Sousa JC, Correia-Neves M, Sousa N, Palha JA, Marques F. The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer's disease. Brain Behav Immun. 2015 Oct;49:280-92. Epub 2015 Jun 16 PubMed.
Stanford University Medical School
Stanford University
Yin and co-workers describe a creative, novel approach to try to understand the physiological role of ApoE and its implications for Alzheimer’s and cardiovascular disease. Using ApoE-knockout and humanized ApoE3- and ApoE4-knock-in mouse models, they examined physiological and immunological changes in the choroid plexus. They report increased expression of interferon pathway genes in the choroid plexus of ApoE knockout mice and that this phenotype is reversed with ApoE3 or 4 reconstitution. This is interesting in that aging seems to induce a similar interferon signature in the choroid plexus of wild-type mice, as shown by Baruch and colleagues, and possibly tying the well-known longevity effects of APOE to this mechanism. Additionally, using binding activity assays, Yin reports that ApoE acts as a checkpoint inhibitor of the classical complement cascade by forming a complex with activated C1q. Excitingly, levels of C1q-ApoE complex seem to correlate with pathology and disease in Alzheimer’s patients and with classical complement cascade activation in atherosclerosis. The authors conclude that the C1q-ApoE complex is a new inhibitor of classical complement activity during disease.…More
Overall, this is a very interesting, rigorous, and exciting observation that links ApoE with anti-inflammatory effects and the complement pathway. Activation of the complement system has long been associated with neurodegeneration and Alzheimer’s disease, although this intricate innate immune pathway has likely beneficial and detrimental effects depending on the local environment and the stage of disease. The current study proposes ApoE as an inhibitor of C1q and highlights ApoE as a new regulator for the classical complement pathway in the choroid plexus.
The study opens many exciting new questions and avenues for more research. How does interferon signaling in the choroid plexus relate to ApoE and brain inflammation and aging in general? How exactly does ApoE regulate complement activation in vivo, and how does this relate to the development of Alzheimer disease? It will also be interesting to identify the cell types involved in this response including the origin of ApoE. Importantly, given attempts to lower ApoE protein levels as a therapy for Alzheimer’s disease, it will be important to monitor activity of the complement cascade and consider these new findings.
Institute for Cardiovascular Prevention (IPEK) Klinikum der Universität München (KUM) Ludwig-Maximilians-University (LMU) Munich
We would like to add comments on a few statements raised in the news story:
Christine Skerka is quoted as saying she “thinks the blood-CSF barrier was compromised as well, because the researchers detected immunoglobulins in the ChP.” We need to clarify: We did not study the blood-CSF barrier. The detection of immunoglobulins in the ChP does not directly support the statement that the blood-CSF barrier is compromised. There are fundamental differences between the blood-brain barrier (BBB) and the blood-CSF barrier (Ransohoff et al., 2012; Schwartz et al., 2014; Shechter et al., 2013). It is important to note that tight junctions forming the BBB are expressed by endothelial cells in the brain parenchyma, while tight junctions forming the blood-CSF barrier are expressed by the ChP epithelial cells. In sharp contrast, the endothelial cell monolayer of the ChP does not participate in the formation of the blood-CSF barrier because it is fenestrated, and even large molecules including immunoglobulins can pass them to enter the space between the endothelium of the ChP and the epithelial cells. Immunoglobulin deposits in the space between the fenestrated endothelial cell monolayer and the epithelial cell monolayer of the ChP are likely unrelated to the blood-CSF barrier. Of course, it is conceivable that the blood-CSF barrier may nevertheless be compromised, but our work does not address or support the speculation by Skerka. Following the lead of Bell et al., however, we observed that the BBB in ApoE-/- and of ApoE4 knock-in mice is compromised (Extended Figure 1e of our manuscript; Bell et al., 2012) and that the BBB breakdown in the brain parenchyma is a function of either the absence of ApoE or the presence of ApoE4. Further studies on the blood-CSF barrier are needed to clarify these important issues.…More
Regarding the question of how ApoE isoforms contribute to AD risk based on the data presented in the manuscript, the story quotes Yadong Huang as saying, “That’s the key unresolved question here.” We reported two ApoE4 isoform-specific pro-inflammatory activities in our manuscript (see below points ii, iii). ApoE4-KI mice on normal chow are normolipidemic and do not have lipid deposits in the ChPs, hence, this data does not directly support the speculation that “the isoforms modulate AD risk by differentially influencing complement activators, such as lipids and Aβ,” as Skerka is quoted as saying. Instead, we propose a different scenario:
Based on the above arguments, we propose that the Janus-headed nature of ApoE4 reveal both anti-inflammatory activities by forming the C1q-ApoE complex (hereby inhibiting the classical complement cascade) and pro-inflammatory activities by triggering a strong IFN signature.
We agree with Huang that the relationship between ApoE and inflammation, and the role of inflammation in neurodegeneration, is not clear-cut. Our study does not clarify this issue, and much work needs to be done to better understand this question. These studies need to take into account major parameters of AD including age, the seeding events during early AD stages, inflammation and its role during AD plaque formation, and synaptic pruning (which may be different during distinct disease stages), and many more, including the isoforms of ApoE. Indeed, recent studies by several groups have indicated that ApoE has opposite effects during the different stages of AD development in mice (Huynh et al., 2017; Liu et al., 2017).
We live in exciting times and hopefully are close to uncovering some AD mysteries, but we are not yet there by any account. Our study adds a little mosaic stone to a big picture that is only beginning to be faintly seen in the distance.
References:
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012 May 24;485(7399):512-6. PubMed. Correction.
Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, Roh J, Finn MB, Sullivan PM, Esparza TJ, Stewart FR, Mahan TE, Ulrich JD, Cole T, Holtzman DM. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron. 2017 Dec 6;96(5):1013-1023.e4. PubMed.
Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, Bu G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron. 2017 Dec 6;96(5):1024-1032.e3. PubMed.
Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012 Sep;12(9):623-35. Epub 2012 Aug 20 PubMed.
Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J. 2014 Jan 7;33(1):7-22. Epub 2013 Dec 19 PubMed.
Shechter R, London A, Schwartz M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat Rev Immunol. 2013 Mar;13(3):206-18. PubMed.
University of California, Irvine
This is a very interesting series of experiments, conclusively demonstrating an interaction between C1q and ApoE, with only slight differences in affinity for C1q among the ApoE isoforms. The authors then provide evidence of an ApoE peptide that prevents this interaction and, since it does not include the regions differing between the ApoE isoforms, this supports the demonstrated lack of difference in affinity between C1q and ApoE isoforms. [However, use of a scrambled peptide or reverse sequence for the 139-152 peptide would have provided more definitive conclusions.] These data add to the growing body of observations providing a link between ApoE and C1q, such as early work by Eikelenboom (Zhan et al., 1995) and later Gareth Howell’s group (Soto et al., 2015). The data also show the ability of β-amyloid fibrils and not soluble amyloid to activate complement, which reproduces and supports previously published work from our lab (Jiang et al., 1994) and others (Tacnet-Delorme et al., 2001). …More
In addition and importantly, the data presented demonstrate that a knockdown of C5 synthesis using novel siRNA reduced plaque-associated microglia in a mouse model of AD. These data are consistent with a critical detrimental role of C5a in the development of cognitive loss in mouse models of AD as we published in 2009 and 2017 (Hernandez et al., 2017; Fonseca et al., 2009), and thus supports the potential for therapeutic targeting of C5a or its receptor C5aR1.
However, to avoid confusion and therefore facilitate progress toward effective therapeutic development, it would be helpful to accurately describe the complement components C1q and C1. There is no “inactive” form of C1q, as mentioned in the paper and the Alzforum report above. In the absence of C1r and C1s, or after the dissociation of the activated enzymes C1r and C1s from C1q (which occurs rapidly in blood due to C1 inhibitor), C1q has many activities as described in the review (Thielens et al., 2017). When C1q is complexed with two molecules each of C1r and C1s in C1, C1 is “unactivated”, but "activatable" when referring to its ability to initiate the CCC. (For some readers, inactive can mean misfolded, inhibited, etc.)
In blood/serum, most of the C1q (90 percent) is found in complex with the proenzymes C1r and C1s (Ziccardi and Tschopp, 1982)—this is “unactivated” C1. When this C1 binds to “activators,” i.e., those molecules that constrain C1q to enable C1r and C1s to be permanently cleaved to active enzymes, the activated C1s (of the now “activated C1”) propagates the complement pathway activity by cleaving the next proteins in the cascade as illustrated in Extended Data Fig. 3 in this manuscript (except that C4 is cleaved first, then C2). [In contrast to what is stated in this manuscript, while C1 is slightly more stable when bound to an activator, C1q bound to a target does not recruit C1r and C1s to any greater extent than free C1q.]
Furthermore, Ca++, which is in all biological fluids, including NHS, is required for binding of C1r2C1s2 to C1q, but not for the “conformational change” induced by the activator to which C1q binds. Therefore, the Ca++ dependence of ApoE binding to C1q does not indicate “that ApoE selectively binds to the activated form, but not the inactivated form of C1q” as concluded in this paper, but rather that ApoE may bind to “free” C1q in a Ca++ dependent manner. Further explorations of the binding of ApoE to free C1q versus C1q in complex with C1r and C1s (C1) are warranted to determine the nature of these interactions in serum and tissue environments.
Deborah A. Fraser, California State University, Long Beach, is a co-author of this comment.
References:
Zhan SS, Veerhuis R, Kamphorst W, Eikelenboom P. Distribution of beta amyloid associated proteins in plaques in Alzheimer's disease and in the non-demented elderly. Neurodegeneration. 1995 Sep;4(3):291-7. PubMed.
Soto I, Graham LC, Richter HJ, Simeone SN, Radell JE, Grabowska W, Funkhouser WK, Howell MC, Howell GR. APOE Stabilization by Exercise Prevents Aging Neurovascular Dysfunction and Complement Induction. PLoS Biol. 2015 Oct;13(10):e1002279. Epub 2015 Oct 29 PubMed.
Jiang H, Burdick D, Glabe CG, Cotman CW, Tenner AJ. beta-Amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol. 1994 May 15;152(10):5050-9. PubMed.
Tacnet-Delorme P, Chevallier S, Arlaud GJ. Beta-amyloid fibrils activate the C1 complex of complement under physiological conditions: evidence for a binding site for A beta on the C1q globular regions. J Immunol. 2001 Dec 1;167(11):6374-81. PubMed.
Hernandez MX, Jiang S, Cole TA, Chu SH, Fonseca MI, Fang MJ, Hohsfield LA, Torres MD, Green KN, Wetsel RA, Mortazavi A, Tenner AJ. Prevention of C5aR1 signaling delays microglial inflammatory polarization, favors clearance pathways and suppresses cognitive loss. Mol Neurodegener. 2017 Sep 18;12(1):66. PubMed.
Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, Taylor SM, Woodruff TM, Tenner AJ. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol. 2009 Jul 15;183(2):1375-83. Epub 2009 Jun 26 PubMed.
Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, Tenner AJ. C1q: A fresh look upon an old molecule. Mol Immunol. 2017 Sep;89:73-83. Epub 2017 Jun 7 PubMed.
Ziccardi RJ, Tschopp J. The dissociation properties of native C1. Biochem Biophys Res Commun. 1982 Jul 30;107(2):618-23. PubMed.
University of Central Lancashire
I notice that everyone is largely in agreement with the findings of the authors. All very exciting. I wish to add that the very sites linked with ApoE and complement activation (atherosclerosis, choroid plexus, amyloid-β plaques) are also known to contain pathogenic bacteria and bacterial toxins. Nobody has mentioned their expert opinion on this in relation to the ApoE-C1q checkpoint story. I have worked on ApoE knockout mouse brains and did not find rampant complement activation. These mice were fed on standard chow. In fact, I only found activated complement after confirming entry of bacteria into the brains of ApoE KO mice. Does this mean that complement activation becomes deregulated in ApoE KO mice fed a high fat diet?…More
Make a Comment
To make a comment you must login or register.