Single-Nuclei Multi-omics Spots Wonky Gene Regulation
Quick Links
Many GWAS variants that raise Alzheimer’s disease risk lie in noncoding stretches of DNA. Do they influence transcription? The answer to this question has been hard to come by, but a new approach might help. Scientists led by Nicholas Cochran, Richard Myers, and Lindsay Rizzardi at HudsonAlpha Institute for Biotechnology, Huntsville, Alabama, have correlated open stretches of chromatin to nearby gene expression in AD and in healthy brain cells. In the February 2 Cell Genomics, they reported 320,000 such chromatin-expression pairs, of which 41,000 were specific to AD.
- Single-nuclei multi-omics connects open chromatin to nearby gene expression.
- The analysis also ties in transcription factor changes.
- Two transcription factors may control the bulk of gene expression alterations in neurons and microglia in AD.
The authors believe that the chromatin—a complex of DNA and protein—contains cis-regulatory elements. Further, they found 430 transcription factors that regulate these CREs. Two stood out. In neurons and in microglia, respectively, ZEB1 and MAFB may regulate transcription from as many as half the CREs unique to AD, hinting that ZEB1 and MAFB may spur faulty gene expression.
“This is the first paper of its kind to do 'true' multi-omics that profiles single-cell RNA- and ATAC-Seq from the same nuclei,” Vivek Swarup, University of California, Irvine, told Alzforum. Assay for Transposase-Accessible Chromatin using sequencing (ATAC-Seq) is a technique that deploys a hyperactive transposase to wedge its way into open stretches of chromatin, cleaving and tagging the labile DNA, which can then be sequenced. “Before, it was just theoretical that this dual analysis should give you higher fidelity, more granular data than previous methods, but this paper shows you that it does” said Swarup.
In past studies, researchers divided the pool of nuclei from a tissue sample into two, used one for snRNA-Seq and the other for snATAC-Seq, then tried to correlate expression data with active chromatin data (Jul 2021 news). “You have to do some computational inference to say cell types in one modality are equivalent to cell types in the other, and you lose confidence in the connections by doing that,” Rizzardi explained. Scientists estimate that, at best, this splitting method matches 45 percent of gene expression to chromatin accessibility (Lee et al., 2023).
To improve the correlation, co-first authors Ashlyn Anderson and Brianne Rogers used a single-cell multiome assay sold by 10X Genomics. It uses ATAC-Seq to tag open stretches of chromatin, then follows up with an RNA-Seq step to measure transcripts, all in the same nucleus. “This way, we are highly confident that an ATAC peak exists in a cell where a certain RNA came from,” Rizzardi told Alzforum.
Anderson and Rogers analyzed 105,000 nuclei isolated from cortical tissue taken postmortem from seven AD cases and eight sex-matched controls. All samples came from the NIH NeuroBioBank. Based on gene expression and chromatin accessibility, the nuclei clustered into 36 subtypes of eight cells.
Next, the researchers matched genes to open stretches of chromatin that might regulate their expression. They picked out ATAC peaks within 500 kilobases of all transcription sites and correlated them to up- or downregulated genes. They predicted that these matched ATAC peaks were likely to be cis-regulatory elements. Of 320,000 potential CREs, 67,500 had been identified as regulatory regions in other independent datasets, such as the Genotype-Tissue Expression Project (GTEx) (Cooper et al., 2022; Bryois et al., 2022; Hu et al., 2021). About 41,000 CREs among six cell types were unique to AD.
Did any CREs control genes linked to neurological diseases in GWAS? Of 3,195 single nucleotide polymorphisms previously tied to AD, the scientists spotted 275 within CREs (Sep 2021 news). These included the rs733839 variant at the BIN1 locus, the rs3865444 and rs12459419 mutations in CD33, and the rs10792832 variant at the PICALM locus.
All were within microglial CREs. In fact, the majority of the SNP-toting CREs unique to AD were found within microglia, reinforcing the field’s emerging consensus that these cells drive disease risk. CREs in other cell types harbored variants linked to other disorders, including autism, bipolar disorder, and schizophrenia.
“We are another step closer to understanding how the noncoding genome associates with disease risk,” co-PI Cochran told Alzforum. Yoon-Seong Kim of Rutgers University, New Brunswick, New Jersey, thinks the authors are on the right track. “Without using single-cell and multi-omic analysis, it is hard to investigate how GWAS SNPs relate to disease in brain cells,” he said.
Did any of the CREs explain expression changes seen in AD? Comparing transcriptomes of the AD and control nuclei revealed 911 differentially expressed genes among six cell types. Of these DEGs, a whopping 94 percent appeared to be regulated by an upstream CRE detected in that same cell type. For example, BIN1 was downregulated in AD microglia and sn-ATAC detected no CREs near the locus in AD microglia despite there being six CREs in control microglia. “Control-specific CREs are transcription factor binding sites normally used to regulate gene expression that are not being used in AD,” Rizzardi explained. The loss of these CREs might explain low BIN1 expression in AD, the authors reasoned.
Three-quarters of all CREs lay upstream of upregulated genes. This makes sense, since transcription factors (TFs) that bind CREs normally induce gene expression. To find out which TFs drove expression of which genes from which CREs, the researchers identified trios comprising: CREs identified by snATAC that contain transcription factor binding motifs; expressed TFs that might bind these CREs; and downstream genes that are up- or downregulated (see image below). They found 60,000 such trios, encompassing 430 transcription factors.
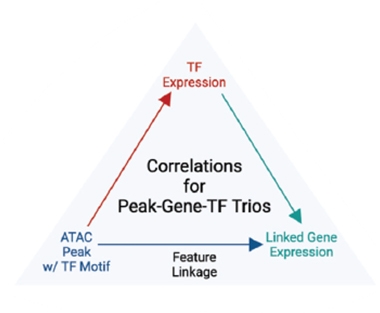
Troublesome Troika? The scientists traced transcription factors that control gene expression by matching stretches of open chromatin containing a TF binding site to that TF's expression and to the genes it regulates. [Courtesy of Anderson et al., Cell Genomics, 2023.]
Of the trios, 1,150 were specific to AD. Among them, two transcription factors stood out. MAFB appeared in 25 percent of the microglial-specific trios, while ZEB1 appeared in half of neuronal trios. Rizzardi was struck by such high enrichment of single TFs. From among these trios, MAFB might regulate the expression of the microglial chemokine receptor CX3CR1 and immune system genes TLR3 and CD84. ZEB1 might control genes involved in ion channel signaling, including the neuronal calcium sensor VSNL1.
Do these trios reflect true regulatory mechanisms? The authors tested this with chromatin immunoprecipitation, which allowed them to see if ZEB1 bound its cognizant regulatory regions. Indeed, the transcription factor latched onto its binding site motif in the CREs of 55 of the 167 neuronal ZEB1 trios tested. “Although the roles of these transcription factors in AD are still unclear, [they] are very promising candidates to be controllers of disease pathology,” wrote Yoshiaki Tanaka of the University of Montreal, Canada.
“Many of the key genes or gene pathways identified in this study recapitulate prior findings in parallel studies, so, in a field with enormous datasets, needles in the haystacks are emerging,” wrote Beth Stutzmann, Rosalind Franklin University of Medicine and Science, North Chicago (comment below). Even so, Stutzmann cautioned, this work is mainly correlative. “It remains to be determined if these gene and transcriptional changes are cause, effect, or a secondary component in AD patients, and it's likely some combination of all three,” she wrote. Still, she noted many of the genes and pathways identified regulate cell functions that can be measured. “Linking gene-level changes with associated changes in function and pathological markers will hopefully be the next step in establishing and differentiating cause and effect at the molecular level,” she added.
Two other studies have used the same multiome platform. One analyzed nuclei from the substantia nigra of Parkinson’s disease cases; the other from cortical tissue of people with C9ORF72 repeat expansions, which cause amyotrophic lateral sclerosis and frontotemporal dementia (Adams et al., 2022; Wang et al., 2023). The former pegged CREs containing 89 PD-associated variants; the latter found CREs unique to early and to late-stage ALS/FTD.—Chelsea Weidman Burke
References
News Citations
- Single-Cell Transcription Cum Chromatin Analysis Pins SREBF1 to AD
- From a Million Samples, GWAS Squeezes Out Seven New Alzheimer's Spots
Paper Citations
- Lee MY, Kaestner KH, Li M. Benchmarking algorithms for joint integration of unpaired and paired single-cell RNA-seq and ATAC-seq data. bioRxiv. February 3, 2023 bioRxiv
- Cooper YA, Teyssier N, Dräger NM, Guo Q, Davis JE, Sattler SM, Yang Z, Patel A, Wu S, Kosuri S, Coppola G, Kampmann M, Geschwind DH. Functional regulatory variants implicate distinct transcriptional networks in dementia. Science. 2022 Aug 19;377(6608):eabi8654. PubMed.
- Bryois J, Calini D, Macnair W, Foo L, Urich E, Ortmann W, Iglesias VA, Selvaraj S, Nutma E, Marzin M, Amor S, Williams A, Castelo-Branco G, Menon V, De Jager P, Malhotra D. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat Neurosci. 2022 Aug;25(8):1104-1112. PubMed.
- Hu B, Won H, Mah W, Park RB, Kassim B, Spiess K, Kozlenkov A, Crowley CA, Pochareddy S, PsychENCODE Consortium, Li Y, Dracheva S, Sestan N, Akbarian S, Geschwind DH. Neuronal and glial 3D chromatin architecture informs the cellular etiology of brain disorders. Nat Commun. 2021 Jun 25;12(1):3968. PubMed.
- Adams L, Song MK, Tanaka Y, Kim YS. Single-nuclei paired multiomic analysis of young, aged, and Parkinson’s disease human midbrain reveals age- and disease-associated glial changes and their contribution to Parkinson’s disease. medRxiv, May 24, 2022. medRxiv
- Wang HV, Veire AM, Gendron TF, Gearing M, Glass JD, Jin P, Corces VG, McEachin ZT. Single nucleus multiome analysis of the prefrontal cortex from C9orf72 ALS/FTD patients illuminates pathways affected during disease progression. bioRxiv. 2023 Jan 13; PubMed.
External Citations
Further Reading
Papers
- Bendl J, Hauberg ME, Girdhar K, Im E, Vicari JM, Rahman S, Fernando MB, Townsley KG, Dong P, Misir R, Kleopoulos SP, Reach SM, Apontes P, Zeng B, Zhang W, Voloudakis G, Brennand KJ, Nixon RA, Haroutunian V, Hoffman GE, Fullard JF, Roussos P. The three-dimensional landscape of cortical chromatin accessibility in Alzheimer's disease. Nat Neurosci. 2022 Oct;25(10):1366-1378. Epub 2022 Sep 28 PubMed.
Primary Papers
- Anderson AG, Rogers BB, Loupe JM, Rodriguez-Nunez I, Roberts SC, White LM, Brazell JN, Bunney WE, Bunney BG, Watson SJ, Cochran JN, Myers RM, Rizzardi LF. Single nucleus multiomics identifies ZEB1 and MAFB as candidate regulators of Alzheimer’s disease-specific cis-regulatory elements. Cell Genomics, February 2, 2023 Cell Genomics
Annotate
To make an annotation you must Login or Register.
Comments
Rosalind Franklin University/The Chicago Medical School
This study brings us closer to zeroing in on the gene-level changes that are associated with AD within specific brain-cell classes by aligning them with corresponding transcriptional regulatory events. This adds an important piece to the puzzle. Many of the key genes, or gene pathways, identified here recapitulate prior findings in parallel studies; so, in a field with enormous datasets, needles in the haystack are emerging.
However, I feel this study suffers from the classic issues of working in postmortem human tissue. It remains to be determined if these gene and transcriptional changes are cause, effect, or a secondary component in AD patients, and it's likely some combination of all three. This doesn't detract from the enhanced confidence this study brings to the growing list, but it cannot address how to interpret these data within the context of pathogenic mechanisms or drivers of disease.…More
Fortunately, many of the critical gene pathways and transcription factors discussed are linked to specific cellular functions that can be measured, so ideally, these RNA-seq and related studies can be accompanied by functional assays measuring calcium handling, synaptic transmission and plasticity, protein mishandling, microglial activation and phagocytosis, and astrocytic signaling in cells derived from these patients, and confirmed using gene-editing toolkits. Linking gene-level changes with associated changes in function and pathological markers will hopefully be the next step in establishing and differentiating cause and effect at the molecular level.
Make a Comment
To make a comment you must login or register.