Profilin 1 Mutant Mouse—a New Model for ALS?
Quick Links
Transgenic animals harboring a mutation linked to amyotrophic lateral sclerosis suffer a similar fate as do humans with the disease, according to new research. In the September 28 issue of Proceedings of the National Academy of Sciences, researchers led by John Landers and Zuoshang Xu of the University of Massachusetts Medical School in Worcester describe how the mice, which express a mutant form of profilin 1 known to cause rare forms of ALS, became weak in adulthood, then paralyzed, and died early. These symptoms were accompanied by the progressive loss of motor neurons in the spinal cord. While aggregates of profilin appeared to gum up the protein degradation machinery in these neurons, the aggregates appeared only after the animals had started displaying symptoms. Researchers hope to use the model to further uncover the cellular mechanisms that underlie ALS.
“Hopefully this PFN1 mouse model will gain traction among ALS researchers similar to the widely used mutant SOD1 mouse, with which it shares several important ALS-like characteristics,” commented Matthew Figley of Washington University in St. Louis, who was not involved in the study. “Much work remains to be done to determine the mechanism of PFN1 motor neuron toxicity but this new mouse model will likely be a vital tool in those pursuits.”
Numerous mouse models based on several other ALS-linked mutations have been recently generated (see Alzforum ALS Research Models). Alas, so far only SOD1 mutant mice, which were first developed more than two decades ago, fully recapitulate the severity of motor neuron degeneration seen in ALS. Since SOD mutations account for a tiny fraction of all ALS cases, researchers have been on the lookout for new models.
Landers’ discovery of ALS mutations in the gene for profilin raised hope, because this actin-binding protein supports the polymerization of the cytoskeleton, which is crucial for axon maintenance and growth (see Jul 2012 news). In mouse neurons and human cell lines, normal profilin comingles with RNA/protein conglomerates called stress granules, and in yeast, ALS-associated mutations in profilin altered stress granule dynamics (see Nov 2013 conference news and Figley et al., 2014). These granules, which harbor another ALS-associated protein, TDP-43, may harm cells when they fail to dissipate and hold important proteins hostage (see Liu-Yesucevitz et al., 2010).
It is currently unclear whether mutant PFN1 exacts its neurodegenerative toll by shirking its actin-binding duties, or consorting with protein aggregates, or both. However, people with PFN1-associated ALS—which occurs in less than 2 percent of people with familial ALS—suffer from many of the hallmark symptoms of the disease, including progressive muscle weakness, paralysis, and early death.
To create a model, first author Chunxing Yang and colleagues generated mice expressing the ALS-associated C71G mutant form of human PFN1. After developing and comparing multiple lines of transgenic mice, the researchers settled on a strain that expressed three copies of the human PFN1 gene—two under control of the Thy1.2 promoter, and one controlled by the mouse prion promoter. Compared with other expression schemes, this triple gene combo yielded the highest level of PFN1 expression in motor neurons and the earliest, most severe ALS symptoms. For controls, the researchers selected a strain of animals that expressed a single copy of wild-type PFN1 at similar levels as the triple combo mutant strain.
For the first three months of their lives, PFN1-C71G transgenic animals appeared the same as nontransgenic animals or those expressing the wild-type version (PFN1-WT) of the protein. However, between 100 and 120 days, about 10 percent of the PFN1-C71G mice started dragging their feet; between 120-140 days, 70 percent did so; and by 170 days, all of these animals limped, some moving slowly. Beyond 170 days, their condition worsened to paralysis of all limbs; at which point the animals could no longer move and were euthanized at an average of 211 days old. Performance on various motor tests, including balancing on a spinning rod, jumping and dangling from the top of their cage, and grip strength, also waned throughout the disease course. In contrast, PFN1-WT animals developed no motor weakness.
Other mouse lines carrying fewer copies of PFN-C71G also developed progressive motor dysfunction and paralysis, but it started later. This indicated that the speed of progression correlated with the amount of mutant PFN1 expressed in neurons, which was fivefold higher than endogenous levels in the triple transgenic.
Examining motor neurons in the ventral horn of the spinal cord, the researchers noticed that between 100 and 120 days (when symptoms first appeared in some mice), axons started degenerating. This ramped up between 120 and 140 days, when neurons started dying and markers of neuroinflammation, including widespread astrogliosis and microgliosis, flared up in the upper reaches of the spinal cord. Some neurons also died in the dorsal root ganglia of the spinal cord as well as the medulla, but not in other parts of the brain.

Profile of Destruction.
By the time animals reached the paralysis stage, motor neurons in the cervical section of the spinal cord were dying in droves in PFN1-C71G mice (right). [Courtesy of Yang et al., PNAS 2016.]
How did this mutation harm motor neurons? The researchers inspected the neurons for signs of flagging profilin function and for protein aggregates. In contrast to cells from PFN1-WT mice, PFN1-C71G motor neurons expressed lower levels of neurofilaments—components of the neuronal cytoskeleton needed for the growth and stability of axons. The network in the mutant neurons looked stunted, forming a circular formation around the nucleus instead of extending a web out to the far ends of axons. Small, to occasionally large, aggregates of PFN1-C71G appeared throughout the cytoplasm. Comingling with the profilin were aggregates of ubiquitin and p62, suggesting there may be malfunctions in the proteasome and autophagy protein degradation pathways, respectively.
Interestingly, the researchers detected no aggregates of PFN1-C71G until day 127, even though axons were degenerating and some mice started dragging their feet between day 100 and 120. More aggregates appeared as the disease progressed. These results indicated that although profilin aggregates worsen with disease severity, they may not initially spark neurodegeneration.
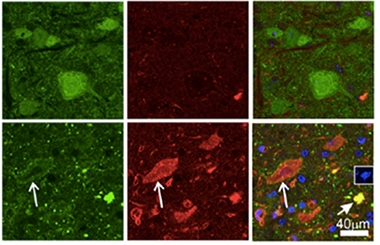
Problem Proteins.
Aggregates of profilin (green) and ubiquitin (red) choked motor neurons in PFN1-C71G mice (bottom) but not in PFN1-WT controls (top). [Courtesy of Yang et al., PNAS 2016.]
How does PFN-C71G kill motor neurons in these mice? And what role, if any, do profilin aggregates might play in the process? Benjamin Wolozin of Boston University School of Medicine proposed that the mutation allows profilin to form oligomers, which interfere with the protein’s role in axonal transport. This could explain axon degeneration prior to the detection of obvious aggregates.
However, Wolozin cautioned that the overexpression of PFN1-C71G makes it difficult to predict whether the same mechanisms would kill neurons in people with ALS. Still, a previous study had indicated that ALS-associated PFN mutations caused the protein to become unstable and prone to aggregation if not quickly degraded (see Jun 2015 news).
Xu agreed that oligomers of profilin could precede the formation of detectable aggregates, however, he hypothesized that profilin mutations led to cytoskeletal disorganization and axon degeneration not by inhibiting profilin function, but by overwhelming the protein quality-control machinery with aggregates. He plans to explore this idea. He also noted that the transgenic animals still expressed normal endogenous profilin, so any phenotype driven by loss of profilin function would need to be a dominant negative one.
Wolozin, who helped characterize TDP-43- and RNA-laden stress granules in ALS, also pointed out that none of these granules accumulate in the PFN1-C71G mice. Xu said they looked for this pathology in vain. While Wolozin praised the model for its strong symptomatic phenotype, he called its lack of TDP-43 pathology disappointing, given that 90 percent of people with ALS have this pathology. This profilin model is therefore unlikely to be helpful in drug discovery studies, he said.
Xu disagreed. He said TDP-43 aggregates are clearly not the only cause of degeneration in motor neurons, and that several mouse models harboring these aggregates lack extensive motor neuron neurodegeneration. Interestingly, in the SOD1 mutant model, which has both severe neurodegeneration but no TDP-43 pathology either, the cytoskeleton crumbles and ubiquitin/p62 aggregates accumulate, just as in the PFN1 mouse, Xu said. Hence, motor neuron death due to overexpression of either mutant PFN1 or mutant SOD1 could be independent of TDP43 pathology, Xu said. He plans to closely compare the two models, and in collaboration with other labs, use the profilin model to test for therapeutics. “At least now we have two models to test,” he said.—Jessica Shugart
References
Research Models Citations
News Citations
- Profilin Gene Is Actin’ in ALS
- Profilin-1 Links Cytoskeleton and RNA Aggregation in ALS
- Mutations Open Cavity in Profilin, Destabilize the ALS-Linked Protein
Paper Citations
- Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci. 2014 Jun 11;34(24):8083-97. PubMed.
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Vanderwyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5(10):e13250. PubMed.
Other Citations
Further Reading
Primary Papers
- Yang C, Danielson EW, Qiao T, Metterville J, Brown RH Jr, Landers JE, Xu Z. Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc Natl Acad Sci U S A. 2016 Oct 11;113(41):E6209-E6218. Epub 2016 Sep 28 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University
This is very nice work from the same team of labs at UMass that first described and characterized the profilin 1 (PFN1) mutations in familial ALS patients just four years ago. Excitingly, they’ve found that high neuronal expression levels of the ALS-causing C71G mutant PFN1, but not similar expression of wild-type PFN1, result in many of the characteristic phenotypes associated with ALS, including adult-onset muscle weakness progressing to paralysis and lethality in their transgenic mice. The authors thoroughly characterized the mutant mice and they show that the ALS-like symptoms are proceeded by prominent axonal degeneration, denervation at neuromuscular junctions, neuroinflammation, and finally drastic spinal cord motor neuron loss. Importantly, the authors also demonstrate that although their transgene and mutant PFN1 protein are highly expressed in other neuron populations (e.g. cortex, hippocampus), the associated cell toxicity is largely confined to the spinal cord motor neurons, with some less severe loss of sensory neurons as well. This selective aspect of their mouse model is critical to future studies of motor neuron susceptibility due to mutant PFN1 expression in ALS.
Yang et al. demonstrate that ubiquitin and p62 are upregulated in motor neurons in the mutant PFN1 mouse, and they propose that this is due to an abnormal proteostasis response. Further investigation into the mutant PFN1 pathology in these mice is warranted. The authors note that the immunostaining for mutant PFN1 shows “small particulates in the cytoplasm and neuronal processes.” These are remarkably similar in appearance to stress granules. Given that we demonstrated PFN1 can itself be a component of stress granules (Figley et al., 2014), it will be interesting to see if these mutant PFN1 aggregates in vivo also contain stress granule proteins, as are often observed in other ALS-related neuropathology. It will also be of interest to test if the mutant PFN1 aggregates contain other known ALS-associated proteins like TDP-43, FUS, or ataxin 2, especially since several groups have now reported mutant PFN1 interacting with TDP-43 (Wu et al., 2012; Tanaka et al., 2016; Matsukawa et al., 2016).
Hopefully, this PFN1 mouse model will gain traction among ALS researchers similar to the widely used mutant SOD1 mouse, with which it shares several important ALS-like characteristics noted above. Much work remains to be done to determine the mechanism of PFN1 motor neuron toxicity, but this new mouse model will likely be a vital tool in those pursuits.
References:
Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci. 2014 Jun 11;34(24):8083-97. PubMed.
Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, Kost JE, Gonzalez-Perez P, Fox AD, Adams J, Taroni F, Tiloca C, Leclerc AL, Chafe SC, Mangroo D, Moore MJ, Zitzewitz JA, Xu ZS, van den Berg LH, Glass JD, Siciliano G, Cirulli ET, Goldstein DB, Salachas F, Meininger V, Rossoll W, Ratti A, Gellera C, Bosco DA, Bassell GJ, Silani V, Drory VE, Brown RH, Landers JE. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012 Aug 23;488(7412):499-503. PubMed.
Tanaka Y, Nonaka T, Suzuki G, Kametani F, Hasegawa M. Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum Mol Genet. 2016 Apr 1;25(7):1420-33. Epub 2016 Jan 28 PubMed.
Matsukawa K, Hashimoto T, Matsumoto T, Ihara R, Chihara T, Miura M, Wakabayashi T, Iwatsubo T. Familial Amyotrophic Lateral Sclerosis-linked Mutations in Profilin 1 Exacerbate TDP-43-induced Degeneration in the Retina of Drosophila melanogaster through an Increase in the Cytoplasmic Localization of TDP-43. J Biol Chem. 2016 Nov 4;291(45):23464-23476. Epub 2016 Sep 15 PubMed.
Make a Comment
To make a comment you must login or register.