Mutations Open Cavity in Profilin, Destabilize the ALS-Linked Protein
Quick Links
Teeth should be free of cavities—and that might go for proteins, too. Cavities appear in profilin as a result of mutations linked to amyotrophic lateral sclerosis, according to a paper in the Proceedings of the National Academy of Sciences USA this week. The gaps seem to destabilize the protein, making it prone to both degradation and aggregation. Filling this space with a small molecule might fortify the mutant profilin and treat this rare genetic form of ALS, suggest the researchers led by Daryl Bosco at the University of Massachusetts Medical School in Worcester.
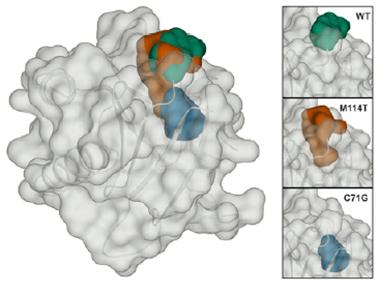
Enlarged cavity:
A small cleft (green) in wild type profilin expands in the M114T mutant (red). Computer models predict the C71G mutation would make another cavity (blue) deeper in the protein. [Image courtesy of Boopathy et al., PNAS.]
Profilin controls the assembly of actin filaments. While a little profilin promotes actin polymerization, a lot shuts it down. A handful of profilin mutations have been found in people with inherited ALS, accounting for an estimated 1 to 2 percent of familial cases. Researchers have not yet worked out how profilin mutations cause ALS. However, the mutant proteins tend to aggregate, suggesting to Bosco that they might adopt abnormal, unstable conformations. (See Jul 2012 news; Smith et al., 2015; Van Blitterswijk et al., 2013).
First author Sivakumar Boopathy investigated the stability of wild-type profilin and four mutants: cysteine-71-glycine, methionine-114-threonine, glycine-118-valine, and glutamate-117-glycine. According to genetics studies, the first three cause ALS, while E117G seems to be a risk factor (Fratta et al., 2014). Boopathy purified the five different recombinant profilins from recombinant E. coli.
To analyze profilin stability, he took advantage of the two naturally fluorescing tryptophans in the protein. The wavelength emitted by these amino acids shifts depending on their immediate environment, which reflects the protein’s conformation. As Boopathy added increasing amounts of urea to denature the proteins, the fluorescence peak shifted from about 354 nanometers to almost 366, indicating that the proteins were becoming less folded. C71G uncoiled at the lowest urea concentrations, followed closely by M114T and G118V. E117G withstood higher levels of the denaturant, unfolding at nearly the same urea concentration as the wild-type protein. This indicated that the trio of M114T, C71G, and G118V were less stably folded than the others. In a separate experiment, the same three mutants had melting temperatures at least 10 degrees lower than the wild-type, again indicating their relative instability, while E117G was off by only a few degrees from wild-type. The unstable triad also migrated more slowly than wild-type on a native protein gel, probably because they were unfolded and took up more space. Overall, the results suggested that the profilin mutants that cause ALS were highly unstable, while E117G was only mildly altered.
Reasoning that the cell should degrade such proteins, Boopathy and colleagues analyzed profilin turnover rates in the human neuroblastoma SKNAS cell line transfected with the different mutants. The researchers first treated the cells with cyclohexamide to halt new protein synthesis, then measured levels of soluble profilin and the insoluble aggregates of C71G, M114T, and G118V. The two pathogenic mutants, C71G and M114T, mostly disappeared within a few hours, suggesting that the proteasome rapidly digested them. The third, G118V, persisted for five hours or more, as did wild-type and E117G profilin. Bosco suspects that malformed profilin can either be directly degraded, or aggregate first. As for G118V, though it was unstable in vitro, cellular factors might have prevented its degradation, the authors speculate.
Next, co-first author Tania Silvas tested the mutants for structural variation. She obtained crystals of the wild type, E117G, and M114T. C71G was insoluble and difficult to purify, yielding an insufficient amount to form crystals, and G118V formed poor crystals. In an X-ray analysis, E117G differed slightly from wild-type, but M114T exhibited a major change, in keeping with its instability data. While the wild-type profilin possesses a small cleft at its surface, this was enlarged in M114T (see image above). Using computer modeling, the authors predicted that the C71G protein would have a cavity in the same region, but the location of glycine 118 in a flexible loop made it hard to predict the mutation’s effect on structure.
The authors conclude that these cavities likely underlie the instability of the three ALS mutations. “I think [the unstable mutants] are misfolded and targeted for degradation,” Bosco said. Exactly what that instability does to motor neurons in people with ALS remains a mystery. “It is possible that [profilin instability] underlies the disease, either through a gain of toxic function or a loss of function,” Bosco said.
What about E117G? “E117G shows a similar trend on protein structure and stability to the pathogenic mutations, but with a more subtle effect. These observations fit exactly with what you would expect from a risk factor,” Julie van der Zee at the VIB University of Antwerp, Belgium, wrote to Alzforum (see full comment below). Van der Zee was not involved in the study.
If these unstable mutants have a hole, the authors reason, then plugging it with a small molecule might help. This kind of “pharmacological chaperone” approach looks promising for other proteins involved in diseases such as cystic fibrosis, noted Gregory Petsko of Weill Cornell Medical College in New York (Leidenheimer and Ryder, 2014; Aymami et al., 2013; Hanrahan et al., 2013). “Anything that will hold the protein’s structure together should work,” Petsko said. Bosco and colleagues are already hunting for small molecules that might shore up the wobbly profilins.
Could such drugs help more than the small group of people carrying those mutations? “You never know,” Petsko said. If profilin participates in ALS-related pathways, stabilizing the wild-type version might be beneficial, he speculated. He suggested testing this question by overexpressing profilin in cells derived from people with ALS.
The question remains: How does unstable profilin instigate ALS? Matthew Figley of Stanford University, who was not involved in the study, wondered about the downstream consequences of unstable profilin. Bosco’s group has already nixed two possibilities, namely that mutants would be unable to bind actin or other binding partners. The mutants bound actin just fine. As for the binding partners, many interface with profilin via a poly-proline motif, and the mutant profilins had no trouble capturing poly-proline in the assays. Bosco plans to look at other profilin-binding proteins and actin dynamics in cells with mutant profilin to figure out its downstream effects.—Amber Dance
References
News Citations
Paper Citations
- Smith BN, Vance C, Scotter EL, Troakes C, Wong CH, Topp S, Maekawa S, King A, Mitchell JC, Lund K, Al-Chalabi A, Ticozzi N, Silani V, Sapp P, Brown RH Jr, Landers JE, Al-Sarraj S, Shaw CE. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol Aging. 2015 Mar;36(3):1602.e17-27. Epub 2014 Oct 31 PubMed.
- van Blitterswijk M, Baker MC, Bieniek KF, Knopman DS, Josephs KA, Boeve B, Caselli R, Wszolek ZK, Petersen R, Graff-Radford NR, Boylan KB, Dickson DW, Rademakers R. Profilin-1 mutations are rare in patients with amyotrophic lateral sclerosis and frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener. 2013 May 2; PubMed.
- Fratta P, Charnock J, Collins T, Devoy A, Howard R, Malaspina A, Orrell R, Sidle K, Clarke J, Shoai M, Lu CH, Hardy J, Plagnol V, Fisher EM. Profilin1 E117G is a moderate risk factor for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2014 May;85(5):506-8. Epub 2013 Dec 5 PubMed.
- Leidenheimer NJ, Ryder KG. Pharmacological chaperoning: a primer on mechanism and pharmacology. Pharmacol Res. 2014 May;83:10-9. Epub 2014 Feb 14 PubMed.
- Aymami J, Barril X, Rodríguez-Pascau L, Martinell M. Pharmacological chaperones for enzyme enhancement therapy in genetic diseases. Pharm Pat Anal. 2013 Jan;2(1):109-24. PubMed.
- Hanrahan JW, Sampson HM, Thomas DY. Novel pharmacological strategies to treat cystic fibrosis. Trends Pharmacol Sci. 2013 Feb;34(2):119-25. PubMed.
Further Reading
Papers
- Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci. 2014 Jun 11;34(24):8083-97. PubMed.
- Chen Y, Zheng ZZ, Huang R, Chen K, Song W, Zhao B, Chen X, Yang Y, Yuan L, Shang HF. PFN1 mutations are rare in Han Chinese populations with amyotrophic lateral sclerosis. Neurobiol Aging. 2013 Jul;34(7):1922.e1-5. PubMed.
- Ingre C, Landers JE, Rizik N, Volk AE, Akimoto C, Birve A, Hübers A, Keagle PJ, Piotrowska K, Press R, Andersen PM, Ludolph AC, Weishaupt JH. A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol Aging. 2012 Nov 8; PubMed.
- Tiloca C, Ticozzi N, Pensato V, Corrado L, Del Bo R, Bertolin C, Fenoglio C, Gagliardi S, Calini D, Lauria G, Castellotti B, Bagarotti A, Corti S, Galimberti D, Cagnin A, Gabelli C, Ranieri M, Ceroni M, Siciliano G, Mazzini L, Cereda C, Scarpini E, Sorarù G, Comi GP, D'Alfonso S, Gellera C, Ratti A, Landers JE, Silani V, . Screening of the PFN1 gene in sporadic amyotrophic lateral sclerosis and in frontotemporal dementia. Neurobiol Aging. 2012 Oct 11; PubMed.
Primary Papers
- Boopathy S, Silvas TV, Tischbein M, Jansen S, Shandilya SM, Zitzewitz JA, Landers JE, Goode BL, Schiffer CA, Bosco DA. Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc Natl Acad Sci U S A. 2015 Jun 30;112(26):7984-9. Epub 2015 Jun 8 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
VIB University of Antwerp
Boopathy et al. provide functional evidence that profilin 1 (PFN1) mutations are pathogenic because they create a cavity in the protein that severely destabilizes it and makes it prone to aggregation, in turn providing mechanistic evidence for the proposed loss-of-function mechanism.
This study is a nice example of how the identification of a rare gene, in this case for ALS, can lead to important mechanistic insights into disease biology, and offer leads for therapy development.
Although initial genetic reports seemed to suggest that PFN1 p.E177G is a benign polymorphism rather than a causal mutation, since it is found in the healthy control population, continued genetic screening in different European populations and meta-analysis showed robust genetic evidence that p.E117G operates as a low-penetrant susceptibility factor that is able to triple ALS risk (Smith et al., 2015). …More
In line with this, Boopathy et al. demonstrate that E117G shows a similar trend on protein structure and stability to the pathogenic mutations, but with a more subtle effect, somewhere between the wild-type and causal mutations, yet closer to the wild-type. These observations fit exactly with what you would expect from a risk factor, and likely E177G leads to disease in conjunction with other genetic and/or epigenetic factors that together lower the threshold for developing ALS.
There is now good statistical evidence that PFN1 p.E177G is genetically associated with ALS and able to significantly increase risk to develop disease. Overall frequencies show an average prevalence of 0.3 percent in patients versus just 0.1 percent in controls. Moreover, the observation by Smith et al. of E177G in one index patient with disease onset of 62 years, an unaffected mother who died at age 62, and a maternal first cousin who died of ALS at age 40, is suggestive for co-segregation of the mutation with reduced penetrance. Also the E117 residue is fully conserved in mammals.
Taken together, I would classify E177G as a low-frequency variant able to cause disease, but with reduced penetrance.
References:
Smith BN, Vance C, Scotter EL, Troakes C, Wong CH, Topp S, Maekawa S, King A, Mitchell JC, Lund K, Al-Chalabi A, Ticozzi N, Silani V, Sapp P, Brown RH Jr, Landers JE, Al-Sarraj S, Shaw CE. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol Aging. 2015 Mar;36(3):1602.e17-27. Epub 2014 Oct 31 PubMed.
Uniformed Services University of the Health Sciences
Interesting study. We had shown a few years ago that mutant huntingtin protein also increased turnover of profilin. Restoring profilin levels was sufficient to improve the disease phenotype. Of course back then we had no clue of the downstream effects of profilin loss outside of altered actin dynamics. Looks like we should dust off those old reagents and take another look at what regulates profilin turnover.
References:
Burnett BG, Andrews J, Ranganathan S, Fischbeck KH, Di Prospero NA. Expression of expanded polyglutamine targets profilin for degradation and alters actin dynamics. Neurobiol Dis. 2008 Jun;30(3):365-74. Epub 2008 Mar 6 PubMed.
Make a Comment
To make a comment you must login or register.