Patricidal Protein? Aβ42 said to Inhibit Its Parent, γ-Secretase
Quick Links
Neuritic plaques, tau phosphorylation, microgliosis—of all the ways Aβ42 could doom the brain, few scientists might have suspected that it cripples γ-secretase, the very enzyme that creates it. Yet that is just what monomers do, according to a manuscript uploaded to bioRxiv. In it, scientists led by Lucia Chávez-Guttiérez, VIB Leuven, Belgium, and William Mobley, University of California, San Diego, report that human Aβ42, though not the mouse version or smaller amyloidogenic peptides, impede processing both of amyloid precursor protein (APP) and other γ-secretase substrates, including Notch, p75, and cadherins.
- In endosomes, Aβ42 can reach micromolar concentrations.
- This inhibits γ-secretase cleavage of APP, p75, and other substrates.
- Modulators could hold Aβ at the secretase active site for full processing.
- One heads to Phase 2 next year.
“It is certainly an unexpected finding,” Harald Steiner, Ludwig-Maximilians-University of Munich, told Alzforum.
Biochemists have studied enzyme inhibition by their products for decades, but whether the example reported here has physiological relevance is unknown. On one hand, it might keep Aβ42 levels from creeping inexorably higher. On the other, it might exacerbate dementia by scuppering the proteolysis of up to 50 different substrates. Colin Masters, The Florey Institute, University of Melbourne, called that a novel hypothesis. Chávez-Guttiérez acknowledged that this will be difficult to prove. “We will try to design experiments to investigate this in the brain,” she told Alzforum. She hopes other scientists will be intrigued enough to investigate the question as well.
In a separate paper published October 18 in The EMBO Journal, Chávez-Guttiérez and colleagues offered a related twist, about γ-secretase’s intramembraneous snipping of successive tripeptides from Aβ. The authors propose that the peptide itself dictates this well-known processivity, not the enzyme. They suggest a tug-of-war between Aβ's hydrophilic N terminal, which pulls the peptide out of the membrane, and ionic interactions at Aβ's hydrophobic C terminal, which pull it into the active site. When the former wins, it rips the substrate out of the membrane too soon, and longer Aβ peptides prevail (image below). When the latter is strengthened—either by way of mutation or small molecule—the substrate lingers longer in the membrane, enabling processing down to short peptides, such as Aβ37, Aβ38, even Aβ26.
“These findings assign a pivotal role to the substrate … in the sequential proteolysis by γ-secretases and suggest it as a sweet spot for the potential design of APP-targeting compounds selectively promoting its processing,” wrote the authors.
Indeed, at this year’s CTAD meeting, held Oct 24-27 in Boston, Agnes Portron of F. Hoffmann-La Roche, Basel, Switzerland, reported that RG6289 may work in just such a fashion. It reduced production of Aβ42 and Aβ40 in favor of Aβ37 and Aβ38, all without affecting the total production of Aβ peptides. Portron reviewed data from a Phase 1 trial of the GSM in healthy volunteers. Roche plans to begin a Phase 2 trial early next year (Nov 2023 conference news).

Aβ Tug o’ War. In this model, inward and outward forces on Aβ48/49 dictate the extent of proteolytic processing happening inside the membrane. If hydrophilic forces yank the peptide out too soon, it retains more amino acids. Modulators that stabilize enzyme-substrate binding might support processivity, leading to shorter peptides. [Courtesy of Koch et al., EMBO J 2023.]
Product Inhibition
First, for readers who'd like a refresher on APP processing: C-terminal fragments (CTFs) are cleaved off APP by β-secretase, then processed by γ-secretase, first by endoproteolysis to release the APP intracellular domain, and then by sequential carboxypeptidase trimming of Aβ peptides from 48 or 49 amino acids to 37 or 38.
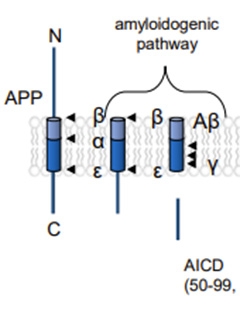
Processing Primer. Sequential endoproteolytic snips of the transmembrane amyloid precursor protein by β- and γ-secretases release the soluble N-terminal domain (top left) and the APP intracellular domain (bottom right). Processive proteolysis by γ-secretase then chops A β's transmembrane domain before it can escape the lipid bilayer. [Courtesy of Zoltowska et al., bioRxiv 2023.]
The idea that Aβ42 might stymie γ-secretase came from a prior work in Chávez-Guttiérez’s lab. Maria Szaruga and colleagues had found that Aβ42 competes with APP C-terminal fragments for binding to γ-secretase (Szaruga et al., 2017). If Aβ42 competes with CTFs, might it prevent them from being processed? Mobley and Chávez-Guttiérez collaborated to find out. Katarzyna Marta Zoltowska in Leuven measured γ-secretase kinetics in cell-free systems and mouse primary neurons, while Utpal Das in San Diego looked for signs of inhibition in cultured human neurons.
Zoltowska found that human Aβ42 curtailed γ-secretase endoprotease activity, tanking production of AICD. Kinetic analysis pointed toward a competitive type of inhibition. The more APP CTF was available, the more Aβ42 was needed to prevent proteolysis. Das found that Aβ42 affected γ-secretase in human neuroblastoma and neural progenitor cells equally. Because of the competitive, and reversible, nature of this inhibition, Chávez-Guttiérez thinks it probably cycles on and off as Aβ42 waxes and wanes.
“We speculate that the alternating mechanism may desynchronize biology and contribute to neuronal toxicity,” Chávez-Guttiérez said. She sees this as secondary to originating pathologies in familial or late-onset sporadic AD. “Those primary pathogenic changes in Aβ production and/or clearance would converge at this secondary mechanism to partially inhibit γ-secretase activity. Then, the latter could lead to altered γ-secretase-mediated signaling events," she said. Indeed, Zoltowska and colleague only saw inhibition at micromolar concentrations of Aβ.
The upshot of this would be a surplus of not only APP CTFs, but also some other γ-secretase substrates, of which there are at least 149 (Güner and Lichtenthaler, 2020). For example, in cells and cell-free assays, the Notch intracellular domain accumulated, as did neurexin, p75, and cadherin substrates.
In this respect, Aβ42 acted akin to γ-secretase inhibitors, paradoxical as that may sound. Mothballed years ago for having sped up rather than slowed cognitive decline in AD trials, GSIs can poison the human immune, intestinal, and neurological systems (Aug 2010 news; Aug 2011 news; Dec 2012 news).
Much of that was initially attributed to interference with Notch signaling, but scientists now think many other substrates got hit, too. Indeed, Chávez-Guttiérez linked the death of cholinergic neurons to GSIs stalling p75 processing. Counterintuitively once again, this neurotrophin receptor causes cell death when it accumulates and oligomerizes on the cell surface (Franco et al., 2021). Zoltowska and colleagues found that Aβ42 mimics this GSI effect on p75 in rat neurons, causing apoptosis driven by caspase 3.
“These observations … are consistent with studies demonstrating that Aβ-induced neuronal degeneration is dependent on the presence of p75NTR,” wrote Frank Longo, Stanford University, California. Longo has long studied this phenomenon and is trying to develop a drug based on it (see LM11A-31).
Besides limiting γ-secretase processing of a plethora of substrates, Aβ42 also led to a build-up of β-CTFs in human neuroblastoma and iPSC-derived neurons. Some scientists, most prominently Ralph Nixon at the Nathan Kline Institute, New York, call these peptides a bigger problem than Aβ42, saying they block vacuolar proton pumps in lysosomes and back up protein degradation.
Nixon’s group wondered about the high concentration of Aβ needed to temper the secretase. “While we question the physiological relevance of the report’s in vitro evidence …, we view contributions to APP β-CTF build-up in neurons by any means to be compatible with our findings,” wrote Juhyun Lee, Ying Jiang, and Nixon (comment below).
Others echoed the concern about the concentration needed to see this effect. “We all know that Aβ can do strange things at micromolar concentrations,” noted Masters. “I would have liked to see more data on specificity, using control peptides,” he wrote. Chávez-Guttiérez believes micromolar concentrations are indeed reached in certain cellular compartments. Others have reported as much in endosomes, and in synapses where enough Aβ accumulates for it to oligomerize (Hu et al., 2009; Pickett et al., 2016). When Zoltowska and colleagues added 2.5 μM Aβ to mouse synaptosomes, β-CTFs accumulated there.
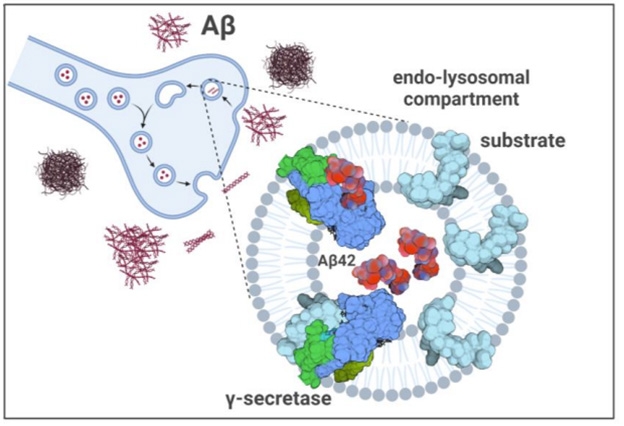
Poisoned Synapse? If Aβ42 (red) reaches micromolar concentrations in synapses, then γ-secretase (blue/green) processing of substrates (cyan) could collapse. [Courtesy of Zoltowska et al., bioRxiv, 2023.]
Tweak versus Block
For their paper in EMBO J, first author Matthias Koch and colleagues used mutagenesis to test if amino acids in the Aβ sequence influence how γ-secretase truncates it.
First, they messed with Aβ amino acids 17 to 32, which protrude just outside of the transmembrane domain. Lysine 28 lies at the membrane, glutamate 22 and aspartate 23 lie a little further upstream; all three seemed to wield particularly strong influence. Hydrophobic substitutions at these positions dramatically revved protease processivity in cells, boosting Aβ37 and Aβ38, at the expense of Aβ40 and Aβ42, by 15-fold. Hydrophobic substitutions at lysine 28 yielded shorter Aβ peptides even when the scientists raised the temperature or added detergent; this suggested to them that the additional hydrophobicity strengthened enzyme-substrate binding, which is typically weakened by such treatments.
What does this say about γ-secretase processing of Aβ? A clue came from making multiple amino acid changes simultaneously. As the number of hydrophobic changes grew, γ-secretase chopped Aβ into ever shorter pieces. For example, when Koch swapped in valine at positions 25-28, γ-secretase mostly churned out Aβ26, a peptide not typically seen under physiological conditions. In effect, Aβ was being sucked further and further into the membrane and into the maw of the enzyme.
These observations prompted the tug-of-war hypothesis—that the hydrophilic N-terminal tries to pull Aβ out of the membrane, while the secretase tries to pull it in by its carboxy terminal. Replacing hydrophilic amino acids there with hydrophobic ones lets the peptide slide further in, the data suggest.
Michael Wolfe, University of Kansas, Lawrence, is unconvinced. In his hands, a carboxyl group at the C-terminal end was not needed for snipping up Aβ. Incorporating an amide group at its position 49 did not impede processing (Fernandez et al., 2016). “As the Aβ49 C-amide at the C-terminus cannot be deprotonated at biological pH, this demonstrated that a C-terminal negative charge (i.e., carboxylate) plays no role in setting up tripeptide trimming,” he wrote (comment below).
Still, if Chávez-Guttiérez’s interpretation is true, it could explain how familial AD mutations in APP work. Those that add outward force in the tug of war would favor longer Aβ peptides. Indeed, Koch found that adding hydrophobic amino acids to Aβ’s N terminal to weaken its outward pull rescued the effect of several FAD mutations (image below).
Wolfe called this result remarkable, but thinks it's because the active site becomes too mechanically stiff to work efficiently. His work suggests that FAD mutations in presenilin 1, the catalytic subunit of γ-secretase, paralyze the enzyme-substrate (E-S) complex, stalling proteolysis. On September 9, co-first authors Sujan Devkota and Vaishnavi Nagarajan at University of Kansas, and Rui Zhou, Tsinghua University, Beijing, and colleagues uploaded a manuscript to bioRxiv. They had synthesized Aβ transmembrane domain mimetics that catch γ-secretase in the act of catalysis. CryoEM analysis of this transition state then allowed them to simulate proteolysis in silico. This predicted that six different PS1 FAD mutations limited the active site's conformational flexibility. The scientists' conclusions? First, catalysis, which requires conformational rearrangement, will slow. Second, the mutant enzyme binds substrate more, not less, tightly than does the wild type.
Chavez-Guttiérez disagrees, claiming that the in silico simulations do not directly assess E-S stability, and that the two labs do not even agree on the term. “It seems that our respective studies choose distinct semantics to define ‘E-S stability’ … hence we believe the two do not contradict each other,” she wrote. For more detailed arguments on either side, see comments below.

Foiling FAD. Two pathogenic mutations in Aβs transmembrane domain, I45F and T43I, favor production of Aβ42 over Aβ40. Putting hydrophobic amino acids into positions 23, 27, and 28 outside the membrane cuts outward force on the substrate, allowing processing to Aβ38 and Aβ37. [Courtesy of Koch et al., EMBO J 2023.]
The tug of war would explain a paradoxical finding about GSIs, namely that some mimic FAD mutations in allowing production of longer Aβ peptides. By competing for the active site, compounds such as semagacestat and DAPT end up giving Aβ the boot before γ-secretase has had time to chop it down to its short forms. Some scientists believe this might have contributed to cognitive worsening in semagacestat trials (Oct 2017 news).
As a corollary, compounds that retain the peptide in the membrane should give the secretase more opportunity to process it. That is what GSM III, a potent imidazole-based γ-secretase modulator from Janssen, seems to do (Velter et al., 2014). Koch and colleagues found GSMIII nullified the effect of the GSI DAPT, shifting production to Aβ37 at the expense of Aβ40.
Speaking of clinical trials, GSMs appear to be making a cautious comeback. For more on Roche’s GSM, see Part 7 of our CTAD series.—Tom Fagan
References
Therapeutics Citations
News Citations
- Second-Generation γ-Secretase Modulator Heads to Phase 2
- Lilly Halts IDENTITY Trials as Patients Worsen on Secretase Inhibitor
- Paris: Semagacestat Autopsy and Other News of Trial Tribulations
- Drug Company Halts Development of γ-Secretase Inhibitor Avagacestat
- Semagacestat, a Pseudo γ-Secretase Inhibitor?
Paper Citations
- Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017 Jul 27;170(3):443-456.e14. PubMed. Correction.
- Güner G, Lichtenthaler SF. The substrate repertoire of γ-secretase/presenilin. Semin Cell Dev Biol. 2020 Sep;105:27-42. Epub 2020 Jun 29 PubMed.
- Franco ML, García-Carpio I, Comaposada-Baró R, Escribano-Saiz JJ, Chávez-Gutiérrez L, Vilar M. TrkA-mediated endocytosis of p75-CTF prevents cholinergic neuron death upon γ-secretase inhibition. Life Sci Alliance. 2021 Apr;4(4) Print 2021 Apr PubMed.
- Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee JM. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A. 2009 Dec 1;106(48):20324-9. PubMed.
- Pickett EK, Koffie RM, Wegmann S, Henstridge CM, Herrmann AG, Colom-Cadena M, Lleo A, Kay KR, Vaught M, Soberman R, Walsh DM, Hyman BT, Spires-Jones TL. Non-Fibrillar Oligomeric Amyloid-β within Synapses. J Alzheimers Dis. 2016 May 30;53(3):787-800. PubMed.
- Fernandez MA, Biette KM, Dolios G, Seth D, Wang R, Wolfe MS. Transmembrane Substrate Determinants for γ-Secretase Processing of APP CTFβ. Biochemistry. 2016 Oct 11;55(40):5675-5688. Epub 2016 Sep 30 PubMed.
- Velter AI, Bischoff FP, Berthelot D, De Cleyn M, Oehlrich D, Jaroskova L, Macdonald G, Minne G, Pieters S, Rombouts F, Van Brandt S, Van Roosbroeck Y, Surkyn M, Trabanco AA, Tresadern G, Wu T, Borghys H, Mercken M, Masungi C, Gijsen H. Anilinotriazoles as potent gamma secretase modulators. Bioorg Med Chem Lett. 2014 Dec 15;24(24):5805-13. Epub 2014 Oct 18 PubMed.
Further Reading
Primary Papers
- Zoltowska KM, Das U, Lismont S, Enzlein T, Maesako M, Houser MC, Franco ML, Moreira DG, Karachentsev D, Becker A, Hopf C, Vilar M, Berezovska O, Mobley W, Chávez-Gutiérrez L. Alzheimer's disease linked Aβ42 exerts product feedback inhibition on γ-secretase impairing downstream cell signaling. bioRxiv. 2023 Oct 28; PubMed.
- Koch M, Enzlein T, Chen SY, Petit D, Lismont S, Zacharias M, Hopf C, Chávez-Gutiérrez L. APP substrate ectodomain defines amyloid-β peptide length by restraining γ-secretase processivity and facilitating product release. EMBO J. 2023 Dec 1;42(23):e114372. Epub 2023 Oct 18 PubMed.
- Devkota S, Zhou R, Nagarajan V, Maesako M, Do H, Noorani A, Overmeyer C, Bhattarai S, Douglas JT, Saraf A, Miao Y, Ackley BD, Shi Y, Wolfe MS. Alzheimer mutations stabilize synaptotoxic γ-secretase-substrate complexes. 2023 Sep 09 10.1101/2023.09.08.556905 (version 1) bioRxiv.
Follow-On Reading
Papers
- Devkota S, Zhou R, Nagarajan V, Maesako M, Do H, Noorani A, Overmeyer C, Bhattarai S, Douglas JT, Saraf A, Miao Y, Ackley BD, Shi Y, Wolfe MS. Familial Alzheimer mutations stabilize synaptotoxic γ-secretase-substrate complexes. Cell Rep. 2024 Feb 27;43(2):113761. Epub 2024 Feb 13 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Stanford University School of Medicine
This work describes a well-designed series of studies testing the hypothesis that the pathological accumulation of Aβ1-42 competes with other substrates for γ-secretase processing, resulting in the accumulation of unprocessed γ-secretase substrates, including their intracellular C-terminal domain fragments (CTFs) and reduced levels of further processed, soluble intracellular domains (ICDs). Given the intracellular signaling roles of CTFs and ICDs, this accumulation could lead to abnormal and potentially deleterious changes in downstream signaling. Some γ-secretase substrates of note include the amyloid precursor protein (APP), the p75 neurotrophin receptor (p75NTR), and pan-cadherins. Aβ1-42-induced accumulation p75NTR CTF is of interest given its capacity to promote degenerative signaling, particularly in the absence of the p75NTR co-receptor, TrkA.…More
These observations, suggesting that one mechanism by which Aβ42 promotes neuronal degeneration is through p75NTR mechanisms, are consistent with studies demonstrating that Aβ-induced neuronal degeneration is dependent on the presence of p75NTR (Sotthibundhu et al, 2008; Knowles et al., 2009; Wilcock et al., 2015; Andrade-Talavera et al., 2021; Wei et al., 2023).
In the PC12nnr5 cell line (absent TrkA receptor), Aβ1-42- induced accumulation of CTF was associated with an increase in cleaved (activated) caspase 3, a signaling element known to trigger apoptosis of neurons and other cell types. I also note the activation of caspase-3 through p75NTR mechanisms, given the role of caspase-3 in enabling the “eat me” signaling thought to promote microglial mediated pruning of synapses (Nonaka and Nakanishi, 2019).
References:
Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ. Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci. 2008 Apr 9;28(15):3941-6. PubMed.
Knowles JK, Rajadas J, Nguyen TV, Yang T, Lemieux MC, Vander Griend L, Ishikawa C, Massa SM, Wyss-Coray T, Longo FM. The p75 neurotrophin receptor promotes amyloid-beta(1-42)-induced neuritic dystrophy in vitro and in vivo. J Neurosci. 2009 Aug 26;29(34):10627-37. PubMed.
Wilcock DM, Hurban J, Helman AM, Sudduth TL, McCarty KL, Beckett TL, Ferrell JC, Murphy MP, Abner EL, Schmitt FA, Head E. Down syndrome individuals with Alzheimer's disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer's disease. Neurobiol Aging. 2015 Sep;36(9):2468-74. Epub 2015 May 30 PubMed.
Andrade-Talavera Y, Balleza-Tapia H, Dolz-Gaitón P, Chen G, Johansson J, Fisahn A. Ablation of p75NTR signaling strengthens gamma-theta rhythm interaction and counteracts Aβ-induced degradation of neuronal dynamics in mouse hippocampus in vitro. Transl Psychiatry. 2021 Apr 9;11(1):212. PubMed.
Wei Z, Yang C, Feng K, Guo S, Huang Z, Wang Y, Jian C. p75NTR enhances cognitive dysfunction in a mouse Alzheimer's disease model by inhibiting microRNA-210-3p-mediated PCYT2 through activation of NF-κB. Int J Biol Macromol. 2023 Jan 15;225:404-415. Epub 2022 Nov 12 PubMed.
Nonaka S, Nakanishi H. Microglial clearance of focal apoptotic synapses. Neurosci Lett. 2019 Aug 10;707:134317. Epub 2019 Jun 5 PubMed.
Nathan Kline Institute/NYU Langone Med
Nathan S. Kline Institute
New York University School of Medicine/Nathan Kline Institute
Zoltowska et al. propose that Aβ42 inhibition of γ-secretase is the principal basis for lysosome dysfunction and the rise of APP-βCTF levels seen in AD brain. We welcome their appreciation of APP-βCTF’s pathogenic potential in AD and the new investigations on its roles. While we question the physiological relevance of the report’s in vitro evidence, which is based on endocytosis of a high (3 uM) human Aβ42 concentration, we view contributions to APP-βCTF build-up in neurons by any means to be compatible with our findings that the early accumulation of APP-βCTF in neuronal autolysosomes, starting well before extracellular amyloid deposition, is the primary cause of lysosomal dysfunction that drives AD pathogenesis (Lee et al., 2022). …More
This APP-βCTF accumulation in lysosomes disrupts their acidification in varied AD models (Jiang et al., 2019) via a specific and direct binding of APP-βCTF within the vATPase complex, which disrupts its assembly and impedes its proton pumping activity (Im et al., 2023). The resulting lysosomal pH rise in vivo broadly impairs proteolysis and causes massive accumulation of varied substrates, including partially inactivated cathepsins and many other soluble lysosomal components that are not γ-secretase substrates (Lee et al., 2022).
The authors could consider experimentally excluding the likelihood that elevated APP-βCTF levels are the more relevant basis for lysosomal deacidification and dysfunction in AD and FAD models than the proposed partial γ-secretase inhibition by exogenously applied human Aβ42. Supporting our mechanistic framework, we find that vATPase assembly/activity is disrupted in human FAD APP or APP/PSEN1 models and similarly in a Down's syndrome (DS) mouse model where elevated mouse βCTF is responsible for vATPase disruption (Im et al., 2023).
In Zoltowska et al., mouse Aβ42 is inactive as a γ-secretase inhibitor. Inhibiting γ-secretase in DS fibroblasts exacerbates lysosome de-acidification in an APP-βCTF-dependent manner, while at the same time, it lowers Aβ42 levels (Im et al., 2023). Moreover, even though Aβ42 levels are vanishingly low in PSEN1-deleted cells, lysosomal deacidification caused by disrupted vATPase assembly causes extreme lysosomal dysfunction and substrate build-up, which can be reversed by restoring normal lysosomal pH (Lee et al., 2015; Lee et al., 2020).
Finally, in the authors’ studies, vATPase inhibition by bafilomycin prolongs APP-βCTF half-life and raises its levels more than does exogenously delivered micromolar Aβ42, which does not change APP-βCTF half-life. If APP-βCTF's half-life is not changed by the degree of γ-secretase blockage, then the mechanism underlying an APP-βCTF increase is obscure.
In our proposed framework of primary lysosomal impairment in AD pathogenesis, we view lysosomal Aβ accumulation and intraneuronal Aβ formation as contributors to neuronal cell death leading to extracellular cored plaques. These events are consequences of the earlier lysosomal de-acidification induced by APP-βCTF. A possible additional effect of γ-secretase inhibition by Aβ42 on APP-βCTF, if confirmed in vivo, would be compatible with our framework.
A pathogenic role of APP-βCTF in driving the endosomal dysfunction that underlies cholinergic neurodegeneration is established and independent of Aβ (refs. in Jiang et al. 2022). Notably, mice expressing rab5 or APPL1 (Kim et al., 2015) to model the immediate downstream signaling by pathological APP- βCTF on endosomes do not raise APP-βCTF or Aβ levels above baseline, yet they phenocopy the basal forebrain cholinergic neurodegeneration seen in DS mouse model, which is dependent on APP-βCTF elevation (Pensalfini et al., 2020; and ms in preparation).
References:
Lee JH, Yang DS, Goulbourne CN, Im E, Stavrides P, Pensalfini A, Chan H, Bouchet-Marquis C, Bleiwas C, Berg MJ, Huo C, Peddy J, Pawlik M, Levy E, Rao M, Staufenbiel M, Nixon RA. Faulty autolysosome acidification in Alzheimer's disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci. 2022 Jun;25(6):688-701. Epub 2022 Jun 2 PubMed.
Jiang Y, Sato Y, Im E, Berg M, Bordi M, Darji S, Kumar A, Mohan PS, Bandyopadhyay U, Diaz A, Cuervo AM, Nixon RA. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99). J Neurosci. 2019 Jul 3;39(27):5255-5268. Epub 2019 May 1 PubMed.
Im E, Jiang Y, Stavrides PH, Darji S, Erdjument-Bromage H, Neubert TA, Choi JY, Wegiel J, Lee JH, Nixon RA. Lysosomal dysfunction in Down syndrome and Alzheimer mouse models is caused by v-ATPase inhibition by Tyr682-phosphorylated APP βCTF. Sci Adv. 2023 Jul 28;9(30):eadg1925. Epub 2023 Jul 26 PubMed.
Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, Mitchell CH, Lloyd-Evans E, Nixon RA. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015 Sep 1;12(9):1430-44. Epub 2015 Aug 20 PubMed.
Lee JH, Wolfe DM, Darji S, McBrayer MK, Colacurcio DJ, Kumar A, Stavrides P, Mohan PS, Nixon RA. β2-adrenergic Agonists Rescue Lysosome Acidification and Function in PSEN1 Deficiency by Reversing Defective ER-to-lysosome Delivery of ClC-7. J Mol Biol. 2020 Apr 3;432(8):2633-2650. Epub 2020 Feb 24 PubMed.
Jiang Y, Rigoglioso A, Peterhoff CM, Pawlik M, Sato Y, Bleiwas C, Stavrides P, Smiley JF, Ginsberg SD, Mathews PM, Levy E, Nixon RA. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: role of APP-CTF. Neurobiol Aging. 2016 Mar;39:90-8. Epub 2015 Dec 2 PubMed.
Jiang Y, Alam JJ, Gomperts SN, Maruff P, Lemstra AW, Germann UA, Stavrides PH, Darji S, Malampati S, Peddy J, Bleiwas C, Pawlik M, Pensalfini A, Yang DS, Subbanna S, Basavarajappa BS, Smiley JF, Gardner A, Blackburn K, Chu HM, Prins ND, Teunissen CE, Harrison JE, Scheltens P, Nixon RA. Preclinical and randomized clinical evaluation of the p38α kinase inhibitor neflamapimod for basal forebrain cholinergic degeneration. Nat Commun. 2022 Sep 21;13(1):5308. PubMed.
Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A, Jiang Y, Nixon RA. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer's disease. Mol Psychiatry. 2015 Jul 21; PubMed.
Pensalfini A, Kim S, Subbanna S, Bleiwas C, Goulbourne CN, Stavrides PH, Jiang Y, Lee JH, Darji S, Pawlik M, Huo C, Peddy J, Berg MJ, Smiley JF, Basavarajappa BS, Nixon RA. Endosomal Dysfunction Induced by Directly Overactivating Rab5 Recapitulates Prodromal and Neurodegenerative Features of Alzheimer's Disease. Cell Rep. 2020 Nov 24;33(8):108420. PubMed.
University of Kansas
This is an interesting follow-up study to Szaruga et al., 2017, from the Chávez-Gutiérrez lab, in which it was first suggested that FAD mutations lead to destabilization of γ-secretase-C99 enzyme-substrate (E-S) complexes. However, neither the present EMBO Journal report nor the 2017 Cell study contain experiments that directly address E-S complex stability. Instead, the papers offer temperature-dependent activity assays measuring the relative proportions of Aβ peptide variants. The finding of increased proportions of longer Aβ peptides with increased temperature is taken as evidence of E-S destabilization. Lower temperatures are sufficient for longer-Aβ proportions from FAD-mutant E-S complexes versus wild-type E-S complexes (Szaruga et al., 2017).…More
In contrast, even with higher temperatures, the proportion of longer Aβ from E-S complexes with artificial mutations in the C99 extracellular domain remains low, suggesting greater E-S stability and more efficient processing (Koch et al., 2023). However, other explanations are possible, and some direct means of detecting E-S complex stability is needed to address what remains a speculation based on activity assays.
We had conducted the same temperature-dependent activity assays 12 years ago (Quintero-Monzon et al., 2011) and refrained from interpreting the results to speculate that increased temperature results in destabilization of E-S complexes. Such speculation was difficult to reconcile with the substantial (threefold) increased proteolytic activity with increasing enzyme reaction temperature from 35 to 45oC in our hands, similar to what Szaruga et al. observed.
Having said that, the new findings that hydrophobic mutations in the C99 extracellular domain allow trimming to very short Aβ peptide products is remarkable. These mutations may, as the authors suggest, facilitate the repositioning of Aβ intermediates to set up for subsequent trimming. The “tug-of-war” model to explain effects of FAD mutations and the artificial C99 ectodomain mutations, however, incorrectly suggests that a negatively charged C-terminal carboxylate of Aβ intermediates (e.g., Aβ49) is necessary for the transition, and that this leads to movement toward positively charged R377 and K380 on presenilin-1. This is incorrect for two reasons. First, the authors cite Bhattarai et al., 2022, to suggest that the negatively charged C-terminus of Aβ49 intermediate is needed to set up for the Aβ49-to-Aβ46 trimming step. In that molecular dynamics study, we found that the presence of protonated, positively charged N-terminus of APP intracellular domain (AICD, coproduct of Aβ49 formation) was essential to set up the Aβ49-to-Aβ46 trimming step. As long as this positively charged AICD was present during the molecular dynamics simulations, either unprotonated, charged Aβ49 or protonated, uncharged Aβ49 worked equally well. Second, we did the biochemical experiment in Fernandez et al., 2016: Synthetic Aβ49 and synthetic Aβ49 with a C-terminal amide led to similar levels and proportions of Aβ40 and Aβ42 upon incubation with γ-secretase. As the Aβ49 C-amide at the C-terminus cannot be deprotonated at biological pH, this demonstrated that a C-terminal negative charge (i.e., carboxylate) plays no role in setting up tripeptide trimming.
One final point: We now have evidence to the contrary of Szaruga et al. Working with Masato Maesako, Massachusetts General Hospital, Boston, we find that FAD mutations lead to stabilization, not destabilization, of E-S complexes, observing this stabilization through fluorescence lifetime imaging microscopy in whole cells (Devkota et al., 2023). This is consistent with our findings from molecular dynamics simulations (with Yinglong Miao) that FAD-mutant E-S complexes have reduced conformational flexibility.
Such reduced flexibility explains the reduced proteolytic activity observed with FAD mutations: In general, conformational flexibility is critical for enzyme activity. Moreover, the reduced flexibility would make it more difficult for bound C99 substrate or Aβ intermediates to dissociate from the γ-secretase complex, as the substrate or intermediate is surrounded by presenilin subunits. We also developed a C. elegans model system for FAD and showed that stabilization of E-S complexes is sufficient for age-dependent synaptic loss and reduced lifespan. Production of neither Aβ42 specifically, nor Aβ in general, is necessary. We suggest that stalled and stabilized γ-secretase E-S complexes can trigger synaptic degeneration, independently of Aβ production.
References:
Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017 Jul 27;170(3):443-456.e14. PubMed. Correction.
Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS. Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer's disease-causing presenilin mutations. Biochemistry. 2011 Oct 25;50(42):9023-35. Epub 2011 Sep 30 PubMed.
Bhattarai A, Devkota S, Do HN, Wang J, Bhattarai S, Wolfe MS, Miao Y. Mechanism of Tripeptide Trimming of Amyloid β-Peptide 49 by γ-Secretase. J Am Chem Soc. 2022 Apr 13;144(14):6215-6226. Epub 2022 Apr 4 PubMed. Correction.
Fernandez MA, Biette KM, Dolios G, Seth D, Wang R, Wolfe MS. Transmembrane Substrate Determinants for γ-Secretase Processing of APP CTFβ. Biochemistry. 2016 Oct 11;55(40):5675-5688. Epub 2016 Sep 30 PubMed.
Devkota S, Zhou R, Nagarajan V, Maesako M, Do H, Noorani A, Overmeyer C, Bhattarai S, Douglas JT, Saraf A, Miao Y, Ackley BD, Shi Y, Wolfe MS. Alzheimer mutations stabilize synaptotoxic γ-secretase-substrate complexes. 2023 Sep 09 10.1101/2023.09.08.556905 (version 1) bioRxiv.
K.U.Leuven and V.I.B.
I would like to start by highlighting that genetic GSEC inactivation in the adult brain leads to age-dependent neurodegeneration (Saura et al., 2004; Tabuchi et al., 2009; Acx et al., 2017; Bi et al., 2021). These studies provide key evidence supporting that GSEC-mediated proteolysis plays essential roles in neuronal physiology.
The bottom-line question that has sparked debate in recent decades is whether mutation-driven inactivation of GSECs underlies FAD, or whether it is an impaired enzyme processivity (leading to altered Aβ peptide profiles) that causes neurodegeneration in FAD.
To my mind, this debate is settled by the following three observations:
These conclusions derived from rigorous kinetic analyses reported in EMBO J a decade ago (Chavez-Gutierrez et al., 2012). Follow-up reports reiterate this finding and multiple groups have reported consistent effects of pathogenic mutations on GSEC processivity, including Fernandez et al., 2014.
This is a key piece of evidence given that the vast majority of FAD-linked mutation carriers are heterozygous for the mutation. In contrast to rescue of the total GSEC activity, the (mutation-driven) enhanced production of longer Aβ peptides prevails in the heterozygous FAD brains (Szaruga et al., 2015). This is likely explained by the fact that longer peptides are released from the membrane and thus out of reach of the membrane-embedded WT enzyme. This study has been recognized as one of key pieces of evidence resolving the debate regarding FAD pathogenesis in this comment (Wolfe, 2015).
If FAD mutations in the enzyme reduced overall proteolytic activity (as primary mechanism arising from stalled E-S complexes, as suggested by Devkota et al.), how would this (substrate-driven) poisoning of the enzyme cause the characteristic amyloid pathology seen in FAD PSEN1/2? And in APP mutation carriers, would this mechanism be still relevant?
Furthermore, assuming that 50 percent of the total GSEC pool in the FAD brain is mutant, and as suggested by Devkota et al., mutant enzymes are stalled in E-S complexes:
Regarding the molecular mechanism(s) underlying the mutation-driven impairments on GSEC function:
A. Our thermoactivity assays, functionally assessing enzyme-substrate (GSEC-APP/Aβ) complexes during proteolysis in Szaruga et al., 2017, demonstrated that:
B. Our investigations showed that pathogenic PSEN1 variants had a direct impact on the stability of the GSEC complex (Figure S4).
C. Finally, the comprehensive and rigorous studies by Okochi et al. (2013) demonstrate that FAD-mutations in PSEN promote dissociation of mutant E-S complexes, while GSMs lower their dissociation. These data are in line with detrimental effects of pathogenic PSEN1 variants on the stability of E-S interactions.
Regarding the preprint by Devkota et al.:
Theoretical studies, using GaMD simulations to analyze how pathogenic PSEN1 mutations affect GSEC APP endopeptidase cleavage show that most of the six PSEN1 mutations visited the active conformation less frequently. This intriguing observation, however, does not directly assess E-S stability, nor is connected to the thermal stability of E-S interactions during proteolysis that we measured in Szaruga et al, 2020. It seems that our respective studies choose distinct semantics to define "E-S stability." The experimental data behind this term is not connected in Devkota et al and Szaruga et al, hence we believe the two do not contradict each other.
The data showing age-dependent synaptic loss in Caenorhabditis elegans are intriguing and are in line with previous findings pointing the essential role of GSEC function in neuronal physiology Saura et al., 2004; Tabuchi et al., 2009; Acx et al., 2017; Bi et al., 2021). Moreover, the data support C. elegans as a model to study the mechanism that underlies the described pathogenesis. However, I find no evidence to conclude that FAD pathogenesis in patients is independent of Aβ production.
References:
Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004 Apr 8;42(1):23-36. PubMed.
Tabuchi K, Chen G, Südhof TC, Shen J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. J Neurosci. 2009 Jun 3;29(22):7290-301. PubMed.
Acx H, Serneels L, Radaelli E, Muyldermans S, Vincke C, Pepermans E, Müller U, Chávez-Gutiérrez L, De Strooper B. Inactivation of γ-secretases leads to accumulation of substrates and non-Alzheimer neurodegeneration. EMBO Mol Med. 2017 Aug;9(8):1088-1099. PubMed.
Bi HR, Zhou CH, Zhang YZ, Cai XD, Ji MH, Yang JJ, Chen GQ, Hu YM. Neuron-specific deletion of presenilin enhancer2 causes progressive astrogliosis and age-related neurodegeneration in the cortex independent of the Notch signaling. CNS Neurosci Ther. 2021 Feb;27(2):174-185. Epub 2020 Sep 22 PubMed.
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem. 2014 Nov 7;289(45):31043-52. Epub 2014 Sep 19 PubMed.
Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda-Falla D, Lleo A, Ryan NS, Lashley T, Fox NC, Murayama S, Gijsen H, De Strooper B, Chávez-Gutiérrez L. Qualitative changes in human γ-secretase underlie familial Alzheimer's disease. J Exp Med. 2015 Nov 16;212(12):2003-13. Epub 2015 Oct 19 PubMed.
Wolfe MS. Cutting to the chase: How pathogenic mutations cause Alzheimer's. J Exp Med. 2015 Nov 16;212(12):1991. PubMed.
Kosik KS, Muñoz C, Lopez L, Arcila ML, García G, Madrigal L, Moreno S, Ríos Romenets S, Lopez H, Gutierrez M, Langbaum JB, Cho W, Suliman S, Tariot PN, Ho C, Reiman EM, Lopera F. Homozygosity of the autosomal dominant Alzheimer disease presenilin 1 E280A mutation. Neurology. 2015 Jan 13;84(2):206-8. Epub 2014 Dec 3 PubMed.
Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017 Jul 27;170(3):443-456.e14. PubMed. Correction.
Petit D, Fernández SG, Zoltowska KM, Enzlein T, Ryan NS, O'Connor A, Szaruga M, Hill E, Vandenberghe R, Fox NC, Chávez-Gutiérrez L. Aβ profiles generated by Alzheimer's disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Mol Psychiatry. 2022 Jun;27(6):2821-2832. Epub 2022 Apr 1 PubMed.
Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, Takeda M. γ-secretase modulators and presenilin 1 mutants act differently on presenilin/γ-secretase function to cleave Aβ42 and Aβ43. Cell Rep. 2013 Jan 31;3(1):42-51. PubMed.
The University of Adelaide
The manuscript from Lucia Chavez-Guttierez and colleagues may prove to be a milestone in the reconciliation of the idea that Aβ is the pathological agent driving Alzheimer’s disease and the alternative view that, instead, APP’s βCTF is critical for the disease. They present evidence of a negative feedback mechanism whereby production of Aβ by γ-secretase cleavage of βCTF is, itself, inhibited by Aβ. Interestingly, they show that the 42 amino acid residue form of Aβ, Aβ42, is most inhibitory toward γ-secretase while shorter or longer forms appear less potent.
The murine form of Aβ42 was considerably less inhibitory than human Aβ42, consistent with the rapid evolutionary divergence of the murine form relative to the Aβs of other tetrapods (Sharman et al., 2013; Moore et al., 2014). One implication of this is that it is less likely that βCTF-equivalent fragments of γ-secretase’s other >100 substrates will have similar inhibitory effects while, conversely, the authors showed that Aβ42 can suppress γ-secretase cleavage of those other substrates, such as NOTCH1 and cadherins.…More
Another interesting aspect of the work: Cell culture medium conditioned by human iPSC-derived neurons homozygous for the Swedish mutant form of APP (which has increased β-secretase cleavage of APP leading to increased Aβ production [Cai et al., 1993]) was, when applied to PC12 cells, measurably more potent in apparent inhibition of γ-secretase than medium conditioned by equivalent non-mutant neurons. This supports that the γ-secretase-inhibitory action of Aβ occurs at physiologically relevant concentrations.
In the discussion, the authors suggest that the inhibitory action of Aβ42 on γ-secretase may explain how fAD mutations in presenilin genes cause γ-secretase loss-of-function phenotypes according to Shen and Kelleher’s “presenilin hypothesis of Alzheimer's disease” (Shen et al., 2007). Here I disagree, since not all fAD mutations in the presenilin genes caused increased production of Aβ42 (Sun et al., 2017) and the human genetic evidence supports that decreased γ-secretase activity causes the skin disease hidradenitis suppurativa/acne inversa but not fAD (Wang et al., 2010).
This message I take from this work: High Aβ levels can inhibit γ-secretase in a physiologically relevant manner to increase levels of βCTF, with predicted impacts on the function of the lysosome (Jiang et al., 2019; Im et al., 2023) and on mitochondria via mitochondria-associated endoplasmic reticulum membrane-related changes (Pera et al., 2017).
References:
Sharman MJ, Moussavi Nik SH, Chen MM, Ong D, Wijaya L, Laws SM, Taddei K, Newman M, Lardelli M, Martins RN, Verdile G. The Guinea Pig as a Model for Sporadic Alzheimer's Disease (AD): The Impact of Cholesterol Intake on Expression of AD-Related Genes. PLoS One. 2013;8(6):e66235. PubMed.
Moore DB, Gillentine MA, Botezatu NM, Wilson KA, Benson AE, Langeland JA. Asynchronous evolutionary origins of Aβ and BACE1. Mol Biol Evol. 2014 Mar;31(3):696-702. Epub 2013 Dec 19 PubMed.
Cai XD, Golde TE, Younkin SG. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993 Jan 22;259(5094):514-6. PubMed.
Shen J, Kelleher RJ. The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007 Jan 9;104(2):403-9. PubMed.
Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc Natl Acad Sci U S A. 2017 Jan 24;114(4):E476-E485. Epub 2016 Dec 5 PubMed.
Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T, Chen M, Sun M, Shen Y, Zhang X. Gamma-secretase gene mutations in familial acne inversa. Science. 2010 Nov 19;330(6007):1065. PubMed.
Jiang Y, Sato Y, Im E, Berg M, Bordi M, Darji S, Kumar A, Mohan PS, Bandyopadhyay U, Diaz A, Cuervo AM, Nixon RA. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99). J Neurosci. 2019 Jul 3;39(27):5255-5268. Epub 2019 May 1 PubMed.
Im E, Jiang Y, Stavrides PH, Darji S, Erdjument-Bromage H, Neubert TA, Choi JY, Wegiel J, Lee JH, Nixon RA. Lysosomal dysfunction in Down syndrome and Alzheimer mouse models is caused by v-ATPase inhibition by Tyr682-phosphorylated APP βCTF. Sci Adv. 2023 Jul 28;9(30):eadg1925. Epub 2023 Jul 26 PubMed.
Pera M, Larrea D, Guardia-Laguarta C, Montesinos J, Velasco KR, Agrawal RR, Xu Y, Chan RB, Di Paolo G, Mehler MF, Perumal GS, Macaluso FP, Freyberg ZZ, Acin-Perez R, Enriquez JA, Schon EA, Area-Gomez E. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017 Nov 15;36(22):3356-3371. Epub 2017 Oct 10 PubMed.
University of Kansas
My remarks below are in reply to Lucia Chávez-Gutiérrez's comments on Devkota et al. The term “enzyme-substrate (E-S) complex” has a specific biochemical meaning, as does the term “stability” of E-S complexes. The Chávez-Gutiérrez lab clearly claims that FAD mutations destabilize γ-secretase E-S complexes to release long Aβ peptide intermediate substrates: This is in the very title of the Szaruga et al., 2017, report. In contrast, our new report, posted in bioRxiv, shows that FAD mutations stabilize E-S complexes, (Devkota et al., 2023), the opposite of the Chávez-Gutiérrez lab findings. This is not a mere difference in semantics, as suggested by Dr. Chávez-Gutiérrez in her comment above.
Both these claims cannot be true. In neither Szaruga et al., 2017, nor in Koch et al., 2023, was E-S complex stability tested in any way. In our new BioRxiv report, we find, working with Masato Maesako at Massachusetts General Hospital, Boston, that FAD mutations lead to stabilization of E-S complexes, observing this stabilization through fluorescence lifetime imaging microscopy in whole cells. It is incorrect to state that we only conducted molecular dynamics simulations as evidence for E-S stabilization. Only one side in this discussion has conducted experiments to test the stability of E-S complexes, and the evidence supports FAD mutations stabilizing these complexes.…More
I respectfully dispute another assertion in Chávez-Gutiérrez's comments: We are not claiming that the stabilized FAD-mutant E-S complexes simply lead to reduction of overall γ-secretase activity. All our studies on the effects of FAD mutations have shown reduction in one or more proteolytic functions of γ-secretase on APP substrate (Quintero-Monzon et al., 2011; Fernandez et al., 2014; Devkota et al., 2021). In some cases, total Aβ production is quite low, but never zero. However, we have consistently observed that FAD mutations skew the proportion of Aβ products to longer forms that are >45 residues due to reduced carboxypeptidase function. We first reported this phenomenon in 2011 using long, hand-cast gels for SDS-PAGE and western blotting (Quintero-Monzon et al., 2011), and more recently used liquid chromatography with tandem mass spectrometry (a method first reported by the lab of Yasuo Ihara) to conduct comprehensive and quantitative analysis of all proteolytic processing steps by γ-secretase on APP substrate (Devkota et al., 2023). We have cross-validated this method in multiple ways to ensure that the effects we observe due to FAD mutations are accurate.
Typically, the reduced carboxypeptidase function caused by FAD mutations also leads to the increased ratios of Aβ42/Aβ40 widely considered critical to AD pathogenesis. We suggest that elevated Aβ42/Aβ40 is a consequence of stalled, stabilized E-S complexes, but that this elevation may not be the trigger of synaptic loss (i.e., increased Aβ42/Aβ40 may be an epiphenomenon). Reduced conformational flexibility of FAD-mutant E-S complexes, which we observe in our molecular dynamic simulations, leads to decreased proteolysis, particularly carboxypeptidase trimming of long Aβ peptides to shorter forms, thereby increasing Aβ42/Aβ40.
However, in our new transgenic C. elegans model system for FAD, where we co-express APP substrate and PSEN1 in neurons, we find that production of Aβ42 specifically, or Aβ generally, is not required for age-dependent synaptic degeneration, only stabilization of E-S complexes. We suggest that the stalled E-S complexes per se, not inhibition of γ-secretase cleavage of other substrates, triggers this synaptic loss. We suggest this in our new report precisely because of the genetic evidence in humans that dominant mutations leading to nonsense-mediated decay of γ-secretase components (true loss-of-function mutations) cause a hereditary skin disease, not neurodegeneration (Wang et al., 2010).
As such, we favor a gain of toxic function due to stabilized E-S complexes. We provide an alternative to the amyloid hypothesis that accounts for FAD mutations in both APP and presenilins, without requiring amyloid production and without invoking loss of γ-secretase activity per se for synaptic degeneration. Aggregation of Aβ42 (or Aβ43) is not a good thing: Evidence suggests that it can, for instance, lead to overactivation of microglia and neuroinflammation. However, after more than 30 years of the amyloid hypothesis, the cumulative evidence that Aβ is the primary driver of the disease remains unconvincing. Perhaps this is why almost all anti-Aβ therapeutic strategies failed in the clinic, and the recently approved anti-amyloid antibodies only modestly slow the rate of cognitive decline. Ours is a fundamentally new way of thinking about how FAD mutations trigger neurodegeneration and deserves serious consideration, after decades of controversy over the role of amyloid and the unimpressive clinical results with anti-amyloid drug candidates.
References:
Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017 Jul 27;170(3):443-456.e14. PubMed. Correction.
Devkota S, Zhou R, Nagarajan V, Maesako M, Do H, Noorani A, Overmeyer C, Bhattarai S, Douglas JT, Saraf A, Miao Y, Ackley BD, Shi Y, Wolfe MS. Alzheimer mutations stabilize synaptotoxic γ-secretase-substrate complexes. 2023 Sep 09 10.1101/2023.09.08.556905 (version 1) bioRxiv.
Koch M, Enzlein T, Chen SY, Petit D, Lismont S, Zacharias M, Hopf C, Chávez-Gutiérrez L. APP substrate ectodomain defines amyloid-β peptide length by restraining γ-secretase processivity and facilitating product release. EMBO J. 2023 Dec 1;42(23):e114372. Epub 2023 Oct 18 PubMed.
Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS. Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer's disease-causing presenilin mutations. Biochemistry. 2011 Oct 25;50(42):9023-35. Epub 2011 Sep 30 PubMed.
Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem. 2014 Nov 7;289(45):31043-52. Epub 2014 Sep 19 PubMed.
Devkota S, Williams TD, Wolfe MS. Familial Alzheimer's disease mutations in amyloid protein precursor alter proteolysis by γ-secretase to increase amyloid β-peptides of ≥45 residues. J Biol Chem. 2021;296:100281. Epub 2021 Jan 12 PubMed.
Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci. 2009 Oct 14;29(41):13042-52. PubMed.
Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T, Chen M, Sun M, Shen Y, Zhang X. Gamma-secretase gene mutations in familial acne inversa. Science. 2010 Nov 19;330(6007):1065. PubMed.
Massachusetts General Hospital/Harvard Medical School
Here, I respectfully avoid commenting on data as those papers are in the preprint stage. On the other hand, I would like to comment from the viewpoint of one of the very few junior faculties (people sometimes call us one of the “endangered species”) who seek to better understand the biology of γ-secretase and its role in AD:
The enormous amount of hard work that γ-secretase researchers have done for the past three decades has given us a massive quantity of knowledge and research materials to study γ-secretase, which I greatly appreciate. Moreover, these extraordinary studies have allowed delivery of medication(s) in the clinic, which may not be perfect yet but could provide hope and a next step to move up. …More
Still, I believe the entire picture regarding why genetic mutations in presenilins (PSEN1/2), the catalytic subunit of γ-secretase, and APP, one of the substrates of γ-secretase, cause early onset FAD neurodegeneration has not been drawn. We all know that the γ-secretase complex with mutations in PSEN1/2 is “inefficient.” It is still unclear to me how much FAD neurodegeneration caused by inefficient γ-secretase is dependent on, and/or independent of, altered Aβ peptide profiles. I.e., is it either or both? If the answer is both, how much is via dependent and how much via independent mechanisms?
γ-Secretase has a wide variety of substrates. Genetic and pharmacological inhibition of γ-secretase cause detrimental effects, but APP is only the one in which FAD mutations have been discovered so far, and the mutation sites are clustered around the Aβ region; therefore, I currently believe the mechanism(s) dependent on altered Aβ peptide profiles to a certain degree play a role in FAD neurodegeneration (but that may change in the future). We all know that various species of Aβ peptides are generated by γ-secretase, long Aβ (e.g., Aβ42, 43) is accumulated in the AD brain, and their removal can slow the progression of the disease in a statistically significant manner.
However, I cannot find any clear answers in previous publications regarding what role(s) longer Aβ species (i.e., Aβ45, 46, 48, 49), which are supposed to be bound to the γ-secretase within the membrane, play, and whether they are associated with FAD neurodegeneration and, if so, how much. It is also unclear whether shorter Aβ species (i.e., Aβ37, 38, 40) and 3-4 amino acids intermediate peptides, generated in the process of the longer Aβ species trimming by γ-secretase, may play some roles. Thus, my lab is currently seeking to determine the role of shorter Aβ species and the 3-4 amino acids peptides inside the cells. I also very much appreciate the recent progress the field has made since we now know the pivotal roles of C99 and p75CTF.
Another challenge I face (as do others) is understanding how γ-secretase functions in intact/live cells and human disease. I guess this challenge could be in part because of the unique environment in which γ-secretase is located and plays its proteolytic role: the lipid membrane bilayer. In other words, γ-secretase can be dynamically affected by changes in lipid membrane properties in living neurons (and likely in glial cells), which seems to be critical in the process of aging and the disease.
I hope our ongoing, healthy, collaborative, and competitive research approach enables us to better understand γ-secretase biology, and that this will soon help treat people with FAD mutations and some sporadic cases!
Make a Comment
To make a comment you must login or register.