New APP Antibodies Expose Variation in Mouse Models of AD
Quick Links
Two new antibodies specific for human amyloid precursor protein (APP) are on the scene, and researchers have used them to reveal markedly distinct brain expression patterns in AD mouse models. As described July 29 in Aging Cell online, the antibodies recognize the N-terminus of human APP in a region well outside the Aβ sequence, and do not cross-react with APP from other species. A test drive of the antibodies in brain sections from 3xTg, Tg2576, and I5 mice indicated that human APP expression patterns differ significantly between the strains. The researchers, led by Stefan Lichtenthaler and Peer-Hendrik Kuhn at the German Center for Neurodegenerative Diseases in Munich in collaboration with Steffen Roßner of Leipzig University, proposed that such differences could explain variation in key behavioral and neuropathological characteristics. The antibodies are freely available to researchers upon request, Lichtenthaler told Alzforum.
“Now that we have tools, we can look at APP expression patterns and use them as a basis for distinguishing between phenotypes,” Lichtenthaler said.
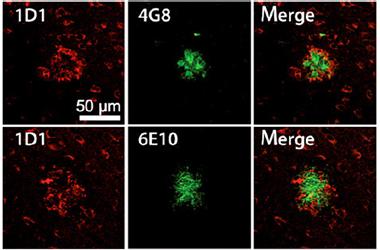
APP Haloes.
Unlike Aβ-specific antibodies 6E10 and 4G8 (green), which bind to the core of plaques, 1D1 detects a halo surrounding the plaques. [Image courtesy of Höfling et al., Aging Cell, 2016.]
Since the discovery that APP can unleash the infamous Aβ peptide, transgenic mouse models expressing the human protein have multiplied. With the exception of recently developed APP knock-in mice, the endogenous mouse APP always lurks in the background. Most of the available antibodies against APP recognize both the human and the mouse proteins, making distinctions between endogenous and transgenic APP difficult. The list of available antibodies is scant. One, P2-1, developed more than 25 years ago when APP was first discovered, fits the bill: It recognizes the N-terminus of APP in a region outside of the Aβ sequence, and only binds to the human protein, but is not widely used currently. Bill van Nostrand, who raised this antibody, told Alzforum that researchers stopped requesting it quite some time ago (see Van Nostrand et al., 1989). P2-1 was once made commercially available, but vendors no longer seem to stock it. Against this backdrop, Lichtenthaler and colleagues wanted additional tools at their disposal to measure human APP expression in AD mouse models, and ultimately to test if differences in these patterns might explain variation in disease characteristics across mouse strains.
Co-first authors Corinna Höfling and Markus Morawski of the University of Leipzig and colleagues raised two rat monoclonal antibodies—dubbed 7H6 and 1D1—against the N-terminal ectodomain of APP. Both antibodies labeled nothing in wild-type mice or rats, but detected human APP in transgenic animals overexpressing the protein. The researchers also confirmed that the 7H6 antibody worked well for flow cytometry, allowing labeling of HEK293 cells overexpressing human APP. On western blots and tissue slices, 7H6 and 1D1 only detected their target under non-reducing conditions, suggesting that the antibodies recognize a conformational epitope held together by a disulfide bond. This contrasts to the anti-APP antibody 22C11, which recognizes N-terminal APP from multiple species under reducing conditions.
That the new antibodies work under non-reducing conditions will allow researchers to examine APP in its native state in the human brain, commented Konrad Beyreuther, who developed the 22C11 antibody back in 1989 (see Weidemann et al., 1989). “Once you denature, you have a whole different world,” Beyreuther said. “Antibodies like these will allow us to focus on the real physiology of APP.” Beyreuther speculates that AD is caused not just by accumulation of Aβ, but also by problems with APP function (see Klein et al., 2016).
Given the new antibodies’ specificity for APP, the researchers wanted to confirm that they would not latch onto Aβ. Indeed, the researchers found that while thioflavin S or Aβ antibodies 6E10 and 4G8 stained the cores of plaques in brain sections of Tg2576 mice, 1D1 produced a halo surrounding the plaques, which Lichtenthaler interpreted as a staining of APP on dystrophic neurites (see image above). 1D1 recognized a similar halo pattern in brain tissue from people with AD, as did the 22C11 antibody.
How would expression patterns of human APP compare among AD mouse models? To find out, the researchers used 22C11 and 1D1 antibodies to label brain sections from 17- to 19-month-old Tg2576, 3xTg, and I5 mice that express wild-type human APP, and wild-type mice. At this age, Tg2576 mice have accumulated plaques in the neocortex, while 3xTg mice have not. I5 mice never develop plaques. The 22C11 antibody detected haloes around plaques in Tg2576 mice; otherwise the binding pattern was the same as in I5 and wild-type mice. The similarities with wild-type mice point to a cross-reaction with mouse APP, said Lichtenthanler. The 3xTg mice also looked similar to wild-type except for strikingly robust binding of 22C11 in layer V pyramidal neurons. In contrast, only brain sections from the transgenic mice, and not from wild-type, bound ID1, in keeping with its specificity for human APP. The new antibody labeled much of the cortex in Tg2576 and I5 mice, but only layer V pyramidal neurons in 3xTg mice. While both 22C11 and 1D1 captured the presence of elevated APP in the layer V pyramidal neurons of 3xTg mice, only 1D1 labeling could definitively link this labeling to the human transgene, as it did not cross-react with mouse APP, noted the authors.
1D1 also revealed that the mouse strains differed in expression of human APP within the hippocampal formation. In I5 mice, CA2/3 and dentate gyrus granule neurons expressed the most hAPP, while in Tg2576 mice, the CA1 to CA3 pyramidal neurons expressed the lion’s share. In 3xTg mice, most of the hippocampal human APP resided in CA1 cells. I5 mice also expressed human APP in the thalamus.
Lichtenthaler did not speculate on how these different expression patterns might affect disease outcomes in the mice, but said the findings should prompt investigators to pay more attention to expression analysis to how results are interpreted.
These expression differences are “less surprising than cautionary, and point to the need to invest in the development of novel mouse models, with targeted replacement of relevant murine genes by their human counterparts,” commented Karen Ashe of the University of Minnesota in Minneapolis (see full comment below and Apr 2014 Alzforum webinar on new APP knock-in mouse models).—Jessica Shugart
References
Research Models Citations
Antibody Citations
Webinar Citations
Paper Citations
- Van Nostrand WE, Wagner SL, Suzuki M, Choi BH, Farrow JS, Geddes JW, Cotman CW, Cunningham DD. Protease nexin-II, a potent antichymotrypsin, shows identity to amyloid beta-protein precursor. Nature. 1989 Oct 12;341(6242):546-9. PubMed.
- Weidemann A, König G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K. Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell. 1989 Apr 7;57(1):115-26. PubMed.
- Klein S, Goldman A, Lee H, Ghahremani S, Bhakta V, UCLA Clinical Genomics Center, Nelson SF, Martinez-Agosto JA. Truncating mutations in APP cause a distinct neurological phenotype. Ann Neurol. 2016 Sep;80(3):456-60. Epub 2016 Aug 4 PubMed.
Further Reading
Primary Papers
- Höfling C, Morawski M, Zeitschel U, Zanier ER, Moschke K, Serdaroglu A, Canneva F, von Hörsten S, De Simoni MG, Forloni G, Jäger C, Kremmer E, Roßner S, Lichtenthaler SF, Kuhn PH. Differential transgene expression patterns in Alzheimer mouse models revealed by novel human amyloid precursor protein-specific antibodies. Aging Cell. 2016 Oct;15(5):953-63. Epub 2016 Jul 29 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Minnesota
This paper very nicely demonstrates that the use of different heterologous promoters to drive various APP cDNAs produces distinct expression patters. This finding is less surprising than cautionary, and points to the need to invest in the development of novel mouse models, with targeted replacement of relevant murine genes by their human counterparts. Not only should specific exons within genes be replaced, but the entire gene, replete with all its regulatory regions, must be carefully excised and the entire human counterpart inserted in its place. Since it is likely that abnormal phenotypes will not arise unless the human genes are over-expressed, the promoter regions may need to be precision-engineered to allow for regulation of expression levels. The technology to accomplish this work exists today but did not, for instance, when I made Tg2576 more than 20 years ago. Unfortunately, we lack funding for such endeavors now. In the meantime, the AD field is, in my opinion, held back by the possibility that the phenotypes in the available mice are not ones that are most relevant to the human disease.
Make a Comment
To make a comment you must login or register.