Does Peripheral Immune Activity Tame Alzheimer’s Disease?
Quick Links
Scientists are still trying to parse out the factors that make inflammation a force for good or ill in Alzheimer’s disease. Now, researchers led by Michal Schwartz at the Weizmann Institute of Science, Rehovot, Israel, suggest it makes a difference whether the immune system is activated peripherally or in the brain. In the August 18 Nature Communications, the authors reported that transiently depleting T regulatory cells in the circulation ameliorated brain pathology and reversed cognitive decline in mouse models of Alzheimer's disease. Because these cells normally quiet immune responses, their depletion released the brakes on systemic inflammation. This, in turn, triggered the choroid plexus in the brain’s ventricles to recruit peripheral immune cells into the brain, the authors found. Inside the brain, those same T regulatory cells that suppressed recruitment in the periphery were beneficial, calming inflammation and helping clear amyloid plaques.
The results suggest that in Alzheimer’s, activation of the peripheral immune response helps fight disease by clearing the way for the body’s own defenses to enter the brain, Schwartz said. It further implies that suppressing systemic immunity would have a negative effect on AD. Calming neuroinflammation, on the other hand, is widely believed to ameliorate neurodegeneration. “We see a nice distinction between how you need to modulate the immune system in the periphery and within brain. This study suggests why systemic anti-inflammatory drugs fell short in Alzheimer’s disease clinical trials,” she told Alzforum.
Other researchers found the data intriguing. “The work convincingly demonstrates that systemic immune suppression can regulate cerebral immune defenses, at least in part by modulating immune cell trafficking to the CNS. This is an exciting and innovative concept in the field,” Maria-Teresa Ferretti at the University of Zurich wrote to Alzforum. At the same time, commenters cautioned that other studies have seen conflicting results from modulating regulatory T cells, saying the big picture may depend on many factors and be more complex than portrayed here.
Evidence is growing that peripheral immune cells may play a role in Alzheimer’s disease. In several studies, monocytes that infiltrate the brain appear to mop up amyloid deposits, however, a recent paper tied the entry of neutrophils to worsening pathology (see Apr 2011 news; Aug 2011 news; Aug 2015 news). In previous work, Schwartz’s group linked brain recruitment of immune cells to expression in the choroid plexus of adhesion factors and chemokines such as ICAM1, VCAM1, and CXCL10. This epithelial membrane inside the brain ventricles separates blood from cerebrospinal fluid. Adhesion factors on the plexus surface capture circulating immune cells and help them squeeze through the membrane. Schwartz and colleagues reported that expression of these adhesion molecules depended on the presence of interferon-γ (IFN-γ) produced by effector memory T cells (see Kunis et al., 2013; Shechter et al., 2013; Aug 2014 news).
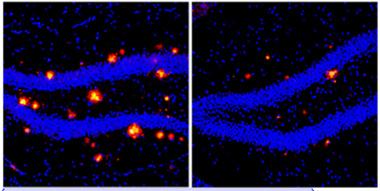
Stronger Immune Response, Less Plaque.
In mice lacking immunosuppressive T regulatory cells (right panel), more immune cells enter brain, and less amyloid (red) accumulates compared to controls (left). [Courtesy of Kuti Baruch and Neta Rosenzweig.]
How might Alzheimer’s disease affect immune cell recruitment? Joint first authors Kuti Baruch and Neta Rosenzweig examined the choroid plexus in 5xFAD mice, which develop aggressive pathology. Starting at 4 months old, these animals expressed only half as many adhesion factors and chemokines in the choroid plexus as age-matched controls, and this correlated with a drop in circulating T cells expressing IFN-γ at the choroid plexus.
The authors wondered if T regulatory cells might mediate this effect. Tregs secrete the cytokine IL-10, which suppresses other immune cells, e.g., expression of IFN-γ by T cells. In keeping with the idea that Tregs slow cell recruitment, the authors saw Treg numbers rise in the 5xFAD mice as disease progressed. To test if Tregs exacerbated pathology, the authors depleted them by crossing 5xFAD mice with animals that expressed diphtheria toxin receptor under the control of a Treg-specific gene promoter. Injecting the offspring with diphtheria toxin selectively depleted just this Treg cell population for a while.
In toxin-treated mice, expression of adhesion factors and chemokines by the choroid plexus soared. Three weeks later the Treg population rebounded. By this point the mice had accumulated twice as many peripheral immune cells, including monocytes and T cells, in their brains as their 5xFAD littermates. Surprisingly, many of these cells expressed Treg markers and secreted IL-10. As the Treg population re-established itself in the treated animals, more of these cells ended up in brain than were seen in controls, the authors note. These Tregs specifically homed to amyloid deposits. In diphtheria toxin-treated mice, plaque burden fell in the cortex and hippocampus (see image above), as did gliosis. The animals performed similarly in the Morris water maze as wild-type. Overall, the data suggest that monocytes and Tregs that enter the brain help to clear plaques and resolve local inflammation, the authors noted.
“These results highlight a need to distinguish between systemic and local contributions of immune suppression in brain pathologies,” Baruch told Alzforum. “It seems counterintuitive, but in order to achieve local suppression of the neuroinflammatory response in neurodegenerative diseases, systemic immunity should be boosted, rather than suppressed.”
Could pharmacological Treg inhibition achieve the same effect as the genetic cross? The authors gave 5-month-old 5xFAD mice weekly injections of glatiramer acetate (GA), an immunosuppressant polymer of four amino acids used to treat multiple sclerosis. This cut Tregs to wild-type levels, correlating with a boost in both choroid plexus adhesion factors and monocyte entry into the brain. Neuroinflammation and plaque load plummeted, and cognition improved to wild-type levels, lasting for a month past treatment.
Tregs inhibit the body’s ability to fight tumors, and are a target in cancer immunotherapy. The authors tried the experimental cancer drug p300i in 10-month-old APPswe/PS1 mice. One week of treatment boosted IFN-γ in the choroid plexus and slashed plaque load up to two months later. Clearance improved with two one-week courses of treatment, separated by a month, and the researchers are currently trying to optimize a protocol in hopes of finding one that may eventually have value in people, Schwartz told Alzforum.
Other researchers cautioned that Treg effects vary in different contexts. For example, Guillaume Dorothee of UMRS 938 Inserm/University Pierre & Marie Curie in Paris presented data at the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18-22 in Nice, France, linking Treg depletion in APPPS1 mice to altered microgliosis and worsening cognition—the opposite of what Schwartz saw in 5xFAD mice (see Apr 2015 conference news). Other work also links infiltrating T cells to accelerating pathology (see Browne et al., 2013; McManus et al., 2014). Moreover, some human data hints that Treg levels fluctuate with disease severity (see Saresella et al., 2010).
“The function of T regulatory cells seems very complex and probably depends on the stage of the disease, neuroinflammatory state, and even strain,” Dorothee told Alzforum.—Madolyn Bowman Rogers
References
News Citations
- CCR2-Positive Macrophages: White Knights of Phagocytes?
- Perivascular Macrophages: The Real Amyloid Clean-Up Crew?
- Could Neutrophils Be the Newest Players in Neurodegenerative Disease?
- Choroid Plexus May Hold a Key To Aging Brain
- Could Adaptive Immunity Set the Brakes on Amyloid?
Research Models Citations
Paper Citations
- Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, Schwartz M. IFN-γ-dependent activation of the brain's choroid plexus for CNS immune surveillance and repair. Brain. 2013 Nov;136(Pt 11):3427-40. Epub 2013 Oct 1 PubMed.
- Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim KW, Klein E, Kalchenko V, Bendel P, Lira SA, Jung S, Schwartz M. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013 Mar 21;38(3):555-69. Epub 2013 Mar 7 PubMed.
- Browne TC, McQuillan K, McManus RM, O'Reilly JA, Mills KH, Lynch MA. IFN-γ Production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease. J Immunol. 2013 Mar 1;190(5):2241-51. PubMed.
- McManus RM, Higgins SC, Mills KH, Lynch MA. Respiratory infection promotes T cell infiltration and amyloid-β deposition in APP/PS1 mice. Neurobiol Aging. 2014 Jan;35(1):109-21. PubMed.
- Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG, Nemni R, Clerici M. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2010;21(3):927-38. PubMed.
Further Reading
Primary Papers
- Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, Tsitsou-Kampeli A, Sarel A, Cahalon L, Schwartz M. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun. 2015 Aug 18;6:7967. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Verona, Italy
This work by Baruch et al. sheds further light on the role of circulating immune cells in the pathogenesis of Alzheimer's disease (AD). The authors found that systemic Foxp3+ regulatory T cells (Tregs) represent a negative player in 5XFAD mice presenting early Aβ pathology by modulating the recruitment of leukocytes to the CNS through the choroid plexus. These results suggest the potential of modulating systemic Treg function as a novel strategy for AD immunotherapy.
A remarkable finding of this study is that one-week treatment inhibiting systemic Tregs led to significant reduction of Aβ accumulation in 5XAD mice during later stages of disease (10 months), which are normally characterized by robust cerebral Aβ plaque pathology. These results are further supported by our recent data showing that timely, short-term therapies that interfere with immune cell function may be beneficial in AD (Zenaro et al., 2015).…More
Treg cells suppress the immune system through antigen-dependent as well as antigen-independent mechanisms. Finding an increase of Treg cells in secondary lymphoid organs in 5XFAD mice is an important observation and it would be interesting to investigate if the peripheral increase of Tregs is antigen-independent or is related to expansion due to CNS or non-CNS antigens.
Previous data have shown that CD4+ regulatory T cells do not only suppress adaptive T cell responses, but are also able to inhibit the functions of monocytes and macrophages in vitro and in vivo (Taams et al., 2005; Maloy et al., 2003). Thus, it would be relevant to further characterize the nature of the interplay between monocytes-macrophages and Tregs that migrated into the brain and seem beneficial in the AD model Baruch et al. used.
The 5XFAD mice display a rapid and aggressive model of Aβ pathology and the relevance of the findings need to be confirmed in other animal models of AD.
References:
Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, Montresor A, Carlucci T, Nanì S, Tosadori G, Calciano L, Catalucci D, Berton G, Bonetti B, Constantin G. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. 2015 Aug;21(8):880-6. Epub 2015 Jul 27 PubMed.
Taams LS, van Amelsfort JM, Tiemessen MM, Jacobs KM, de Jong EC, Akbar AN, Bijlsma JW, Lafeber FP. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum Immunol. 2005 Mar;66(3):222-30. PubMed.
Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003 Jan 6;197(1):111-9. PubMed.
University of Zurich
This work convincingly demonstrates that systemic immune suppression can regulate cerebral immune defenses, at least in part by modulating immune cell trafficking to the central nervous system. This is an exciting and innovative concept in the field. It is a bit counterintuitive that by interfering with T regulatory cells (Tregs) you end up with more immune suppression in the brain, but this only highlights how little we understand the mechanisms of the immune system in brain.
These findings further confirm that the brain is far from being an immune-privileged site, and its homeostasis crucially depends on the proper functioning of the whole immune system. I agree with the authors that modulating the adaptive immune system to induce a beneficial immune response (by peripheral macrophages as well as by microglia) is an attractive therapeutic possibility for AD. …More
To this end, however, it remains to be clarified whether the problem lies in the periphery or at the local level (i.e., infiltrating-patrolling T cells), and how the two systems cross-talk during disease progression. Dysregulation of the adaptive immune system in AD has been proposed, however, results from blood analysis so far have been inconsistent in support (nicely reviewed by Town, 2005). In fact, the evidence for increased Tregs in AD is sparse; on the other hand, a recent paper found even higher activation of CD8 cells in CSF of mild AD patients compared to healthy elderly controls (Lueg et al., 2015). While other work suggests increased activation of infiltrating T cells in brains of aged APP-PS1 mice (Browne et al., 2013), our group has recently observed signs of immunosuppression in the brains of two independent APP-tg mice models in the absence of major systemic changes at very advanced stages of the pathology (15- to 24-month-old mice), thus indicating a local rather than global immunomodulatory effect of Aβ pathology (presented at the last AD/PD, papers under revision). Adaptive immune activation levels most likely change with age and disease progression; whether any of the above preclinical results holds true in AD patients remains to be established.
In summary, the paper from Baruch et al. centered on a crucial and yet overlooked aspect of AD pathology; I hope that its publication will fuel more basic and clinical research to clarify the local and systemic adaptive immune response to AD pathology.
References:
Town T, Tan J, Flavell RA, Mullan M. T-cells in Alzheimer's disease. Neuromolecular Med. 2005;7(3):255-64. PubMed.
Lueg G, Gross CC, Lohmann H, Johnen A, Kemmling A, Deppe M, Groger J, Minnerup J, Wiendl H, Meuth SG, Duning T. Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer's disease. Neurobiol Aging. 2015 Jan;36(1):81-9. Epub 2014 Aug 23 PubMed.
Browne TC, McQuillan K, McManus RM, O'Reilly JA, Mills KH, Lynch MA. IFN-γ Production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease. J Immunol. 2013 Mar 1;190(5):2241-51. PubMed.
Make a Comment
To make a comment you must login or register.