Could Neutrophils Be the Newest Players in Neurodegenerative Disease?
Quick Links
Growing evidence suggests the peripheral immune system plays a role in Alzheimer’s disease. Now, researchers led by Gabriela Constantin at the University of Verona, Italy, turn the spotlight on an overlooked peripheral cell: the neutrophil. These short-lived phagocytes are the most abundant white blood cells, and swarm to sites of inflammation, but have not been thought to act in the brain. However, in the July 27 Nature Medicine, Constantin and colleagues reported that neutrophils seep into the brain in both Alzheimer’s disease and in mouse models of AD. In mice, blocking a neutrophil receptor prevented the cells from entering the brain, and reduced amyloid load and microgliosis, as well as giving the animals a boost in long-term memory. The results highlight the importance of peripheral innate immunity in Alzheimer’s, and hint that neutrophils might be one source of tissue damage.
“This is a thought-provoking study that departs from what everybody else is doing,” Tony Wyss-Coray at Stanford University School of Medicine, California, told Alzforum. “One of the most exciting observations is that the immune response seems most prominent at the early stages of disease in the mouse models.” If the findings hold up, they could point to a potential window for anti-inflammatory therapeutic intervention, he added. Other researchers found the data intriguing as well, but cautioned that more studies will be needed to discover if neutrophil infiltration contributes significantly to human disease.
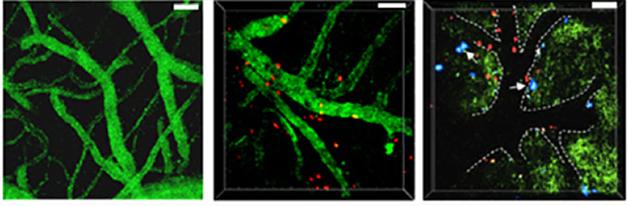
Sneaking Into the Brain. In 5xFAD mice (middle), but not wild-type (left), neutrophils (red) squeeze out of blood vessels (green) and into the brain. Many neutrophils (right) also tarry on blood vessel walls near Aβ deposits (blue). [Courtesy of Nature Medicine, Zenaro et al.]
Several studies report that blood cells such as monocytes and T cells trickle into the AD brain, with monocytes in particular credited with mopping up amyloid deposits (see Apr 2011 news; Aug 2011 news; Apr 2015 conference news). Few researchers have focused on neutrophils, perhaps because these cells live for only hours and would be unlikely to accumulate in tissue. However, one recent study did find neutrophils infiltrating the brain in the 5xFAD mouse (see Baik et al., 2014).
Constantin’s interest grew out of her work studying white blood cell migration in several neuroinflammatory conditions (see Rossi et al., 2011). She was intrigued by the fact that brain endothelial cells in both 5xFAD and 3xTg mice expressed high levels of adhesion factors, suggesting they might capture circulating white blood cells, particularly neutrophils. Joint first authors Elena Zenaro and Enrica Pietronigro found that adhesion molecules were particularly prominent on endothelium near amyloid deposits. Moreover, adding Aβ42 to cultured brain endothelial cells induced expression of cell-surface adhesion factors.
The authors next examined what happened to neutrophils. In brain sections from both mouse models, but not controls, they found the cells loitering outside blood vessels (see image above). They identified them by staining for Ly6G, a cell-surface receptor specific for mouse neutrophils. Accumulation of these cells peaked around the age when cognitive problems began in each model, and occurred in areas with high plaque load and few neurons. This infiltration might have negative consequences, the authors speculated. Neutrophils release many toxic substances, such as reactive oxygen species, cytokines, and neutrophil extracellular traps (NETs), proteins that cage bacteria and damage tissue. The authors detected NETs and the pro-inflammatory cytokine IL-17 around neutrophils in brain tissue, hinting that the phagocytes might be wreaking havoc.
The cells might also injure the blood-brain barrier, the authors suggested. In live imaging experiments, they saw neutrophils crawling along brain blood vessels near amyloid deposits (see image above). Others agreed with this idea. “I find the interaction of the leukocytes at the vasculature as interesting as the extravasation,” Wyss-Coray commented. When white blood cells stick to endothelial cells, they communicate with them through surface receptors, potentially changing the permeability of the endothelium, he noted. Several studies implicate a leaky blood-brain barrier in AD (see May 2014 conference news; Jan 2015 news; Feb 2015 Webinar).
What causes neutrophils to invade the brain? In cell culture experiments, the authors found that synthetic Aβ42 oligomers caused the neutrophil receptor LFA-1 integrin to change shape, assuming a form with high affinity for endothelial adhesion factors. To test the role of this integrin, the authors isolated neutrophils from integrin knockout mice and injected them into 5xFAD animals. The integrin-deficient cells failed to stick to blood vessels, and stayed out of the brain. Likewise, administering an anti-LFA-1 antibody to 5xFAD mice slowed migration of endogenous neutrophils into the brain (see image below).
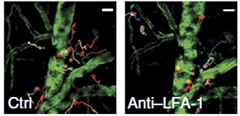
Closing the Door.
Neutrophils (red) in 5xFAD mice creep from blood vessels (green) into brain (left), but remain inside vessels when LFA-1 integrin is blocked (right). [Courtesy of Nature Medicine, Zenaro et al.]
The authors wondered what effect blocking neutrophil entry would have. They injected anti-Ly6G antibodies into 6-month-old 3xTg mice for four weeks, causing neutrophil numbers to crash. Then they allowed neutrophils to recover for four weeks before testing cognition. While the 6-month-old animals had memory problems, the treated 8-month-olds performed like wild-types in the Y maze and in contextual fear conditioning. The cognitive improvements persisted even six months later, the authors reported. In addition, at eight months the treated mice had about a quarter less microgliosis, half as much insoluble Aβ42, and two-thirds less phosphorylated tau than untreated littermates. Meanwhile, levels of the presynaptic protein synaptotagmin rebounded to normal levels. Treatment with other antibodies targeted to neutrophils, such as anti-LFA-1, produced the same effects. So did crossing 3xTg mice with the integrin knockouts.
“This is the first demonstration that neutrophils are important in [Alzheimer's] disease pathogenesis,” Constantin told Alzforum. The long-term improvements struck her. “We think neutrophils are particularly important in the early phases of disease, during which inflammation mechanisms predominate. Intervening during those early phases could be beneficial.”
Does this happen in human disease? The authors stained postmortem brain sections from 11 AD patients and 11 controls, and saw about 10 times more neutrophils in the brain tissue from patients. The cells accumulated near amyloid plaques, as did NETs. As in mice, neutrophils stuck and spread across the walls of brain blood vessels in AD samples, suggesting they could be injuring the blood-brain barrier. While preliminary, the findings appear consistent with a role in Alzheimer’s disease.
Piet Eikelenboom at Vrije University, Amsterdam, pointed out that systemic inflammation in older people can trigger late-onset dementia (see Fong et al., 2015; Mar 2015 conference news). He wondered if blocking neutrophils during a bout of delirium might lower the risk of developing dementia later. Anti-integrin therapies are in use for some autoimmune diseases, and anti-LFA-1 has been in clinical trials, opening the door for testing this approach in people.
At the same time, several researchers said that more work needs to be done to nail down the mechanisms behind the observed improvement in treated mice. Eikelenboom speculated that neutrophils may have their greatest effect on the blood-brain barrier, rather than on parenchyma. Researchers have delineated two stages to crossing the blood-brain barrier: first, crossing the blood vessel wall; second, crossing the glial membrane formed by astrocyte endfeet to enter the brain parenchyma proper. Many white blood cells never make it past the first stage (see Bechmann et al., 2007). It is not clear from the images in the paper whether neutrophils pass this second barrier, Eikelenboom said.
Moreover, Terrence Town at the University of Southern California, Los Angeles, noted that the total number of infiltrating neutrophils in transgenic mice is quite small. Even among those white blood cells that sneak into brain, only about 2 to 3 percent are neutrophils, according to the authors’ measurements. “Is that sufficient to mediate a biological effect?” Town asked. Instead, changes in microglia might account for the improvements in pathology, he speculated. Microglia can express LFA-1 integrin. Perhaps antibodies against this receptor enter the brain and act directly on these cells to dampen inflammation, he suggested.—Madolyn Bowman Rogers
References
News Citations
- CCR2-Positive Macrophages: White Knights of Phagocytes?
- Perivascular Macrophages: The Real Amyloid Clean-Up Crew?
- Could Adaptive Immunity Set the Brakes on Amyloid?
- Fluid Markers and Imaging Back Idea of Breached Blood-Brain Barrier
- In Aging Brain, Blood-Brain Barrier Starts Leaking in Hippocampus
- Systemic Inflammation: A Driver of Neurodegenerative Disease?
Research Models Citations
Webinar Citations
Paper Citations
- Baik SH, Cha MY, Hyun YM, Cho H, Hamza B, Kim DK, Han SH, Choi H, Kim KH, Moon M, Lee J, Kim M, Irimia D, Mook-Jung I. Migration of neutrophils targeting amyloid plaques in Alzheimer's disease mouse model. Neurobiol Aging. 2014 Jun;35(6):1286-92. Epub 2014 Jan 8 PubMed.
- Rossi B, Angiari S, Zenaro E, Budui SL, Constantin G. Vascular inflammation in central nervous system diseases: adhesion receptors controlling leukocyte-endothelial interactions. J Leukoc Biol. 2011 Apr;89(4):539-56. PubMed.
- Fong TG, Davis D, Growdon ME, Albuquerque A, Inouye SK. The interface between delirium and dementia in elderly adults. Lancet Neurol. 2015 Aug;14(8):823-32. Epub 2015 Jun 29 PubMed.
- Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)?. Trends Immunol. 2007 Jan;28(1):5-11. Epub 2006 Nov 30 PubMed.
Other Citations
Further Reading
News
- TREM2 Data Surprise at SfN Annual Meeting
- Inflammation in Midlife May Presage Cognitive Decline
- Neuroinflammation Field Grapples With Complexity at Keystone Symposia
- Innate Immune Cells Enlisted to Clear Amyloid, Fight Disease
- Soothing Neuroinflammation Quells Plaques in Mice
- DC: Is Alzheimer's Rooted in the Early Life?
- Australia Report: Inflammation
Primary Papers
- Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, Montresor A, Carlucci T, Nanì S, Tosadori G, Calciano L, Catalucci D, Berton G, Bonetti B, Constantin G. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. 2015 Aug;21(8):880-6. Epub 2015 Jul 27 PubMed.
Follow-On Reading
Papers
- Rosenzweig N, Kleemann KL, Rust T, Carpenter M, Grucci M, Aronchik M, Brouwer N, Valenbreder I, Cooper-Hohn J, Iyer M, Krishnan RK, Sivanathan KN, Brandão W, Yahya T, Durao A, Yin Z, Chadarevian JP, Properzi MJ, Nowarski R, Davtyan H, Weiner HL, Blurton-Jones M, Yang HS, Eggen BJ, Sperling RA, Butovsky O. Sex-dependent APOE4 neutrophil-microglia interactions drive cognitive impairment in Alzheimer's disease. Nat Med. 2024 Oct;30(10):2990-3003. Epub 2024 Jul 3 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
ImmunoBrain Checkpoint, Ltd.
Weizmann Institute of Science
Brigham and Women's Hospital, Boston
It is becoming increasingly clear that circulating immune cells play a role in Alzheimer’s disease (AD) pathophysiology. These cells can play a direct role in affecting the local neuroinflammatory process associated with AD, as well an indirect role via their peripheral immune activity, which in turn affects disease pathophysiology. Importantly, both protective and destructive roles for CNS-infiltrating immune cells (mainly of the myeloid origin) were documented in AD mouse models, reflecting differences in timing, phenotype, and location of immune-cell activity.
Here, Zenaro et al. report that neutrophils traffic into the brains of AD transgenic mice, starting in early stages of disease, and that transient/partial early neutrophil depletion or LFA-1 blockade during these stages has a beneficial effect, a month later, on disease pathology. It is yet to be determined, however, whether the observed effect on disease pathology is an outcome of inhibiting neutrophil trafficking to the CNS, or rather reflects a peripheral effect on circulating immune cell populations in favor of those that are needed for CNS repair, such as myeloid cell activity/ recruitment to the CNS. As neutrophil infiltration is one of the earliest immunological events in any inflammatory response, it would be also important to find out whether similar intervention would be of benefit if applied at a late progressive stage of the disease, or the benefit is restricted to early disease stages. …More
Inserm / Sorbonne University, Paris, France
This study by Zenaro and colleagues unravels an unexpected instrumental role of neutrophils in the pathophysiology of Alzheimer’s disease (AD). Strong evidence is provided for the recruitment of neutrophils into AD brains, both in murine models of Alzheimer-like pathology and in individuals with AD. Data suggest that Aβ-mediated activation of brain endothelial cells and peripheral neutrophils promotes LFA-1-mediated intravascular adhesion and intraparenchymal motility of neutrophils into AD brains. Depletion of neutrophils or inhibition of their trafficking to the brain improves memory and reduces AD-like pathology in murine models. The therapeutic effect of modulating neutrophils was impressive to me: Transiently interfering with neutrophil response in already cognitively impaired animals restored cognitive functions. Importantly, the beneficial effect lasted for several weeks or months after treatment completion, suggesting a strong impact on the kinetics of disease progression.…More
Neutrophil depletion reduced Aβ load and lowered the amount of phosphorylated tau in 3xTg-AD mice. The extent of microgliosis was also reduced when interfering with the neutrophil response. Regulation of microglial activity has been shown to modulate both Aβ clearance and tau phosphorylation (Heneka et al., 2015); crosstalk may mutually amplify and sustain activation of these innate immune effectors, as suggested by Zenaro and colleagues. On the other hand, structures of amyloid fibrils have been shown to trigger the release of neutrophil extracellular traps (NETs), causing fibril fragmentation into toxic oligomers by NET-associated elastase (Azevedo et al., 2012). These data reported for amyloid fibrils from three different sources (α-synuclein, Sup35, and transthyretin) suggest that NETs may similarly promote the release of toxic soluble Aβ species from amyloid plaques in AD. Further studies will thus be needed to decipher to what extent neutrophils may impact Aβ or tau pathology through modulation of microglial activity, and through direct effector mechanisms, e.g., by NET-mediated processes. Direct effects of neutrophils on neuron survival and activity, as well as on astrocytes, should also be addressed.
In addition to parenchymal trafficking of neutrophils in the cortex and hippocampus, Zenaro and colleagues describe an infiltration of neutrophils in the choroid plexus (CP), which is a selective and "educative" gate of entry for peripheral immune cells into the CNS. T cells have been shown to play a critical role in modulating the functionality of the CP epithelium, thus helping control the recruitment of inflammation-resolving monocytes-derived peripheral macrophages into the inflamed CNS (Schwartz and Baruch, 2014). Of note, neutrophils have been shown to engage into complex bidirectional interactions with not only innate but also adaptive immune effectors, including T cells, leading to reciprocal modulation of their activation status and effector functions (Scapini and Cassatella, 2014). The current work by Zenaro and colleagues raises the interesting hypothesis that neutrophils may participate in modulating the functionality of the CP, and thus its capacity to recruit other peripheral immune populations into the CNS. Such potential effect of neutrophils may rely either on direct interactions with CP epithelium, or its indirect modulation through regulation of CP-infiltrating T cells. Further studies will be needed to address these questions, particularly in the light of recently emerging data suggesting an instrumental role of T cells in the pathophysiology of AD.
This interesting study by Gabriela Constantin’s team highlights neutrophils as new key innate immune effectors to be considered in AD, and opens new perspectives in the development of innovative immunotherapy approaches targeting neutrophils. Importantly, it also further strengthens the need to better understand the complex role of neuroimmune interactions in the pathophysiology of AD, with a particular focus on both peripheral innate and adaptive immune effectors and their interplay with inflamed CNS.
References:
Azevedo EP, Guimarães-Costa AB, Torezani GS, Braga CA, Palhano FL, Kelly JW, Saraiva EM, Foguel D. Amyloid Fibrils Trigger the Release of Neutrophil Extracellular Traps (NETs), Causing Fibril Fragmentation by NET-associated Elastase. J Biol Chem. 2012 Oct 26;287(44):37206-18. PubMed.
Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nat Immunol. 2015 Mar;16(3):229-36. PubMed.
Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood. 2014 Jul 31;124(5):710-9. Epub 2014 Jun 12 PubMed.
Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J. 2014 Jan 7;33(1):7-22. Epub 2013 Dec 19 PubMed.
Vrije Universiteit
Clinical and epidemiological studies in the elderly have shown that events associated with a low-grade systemic inflammatory response (such as hip surgery and mild urinary infection) can lead to both an acute transient cognitive disturbance (delirium) and a long term cognitive impairment (dementia) (Eikelenboom et al., 2012; Fong et al., 2015). Highly relevant for further clinical research is, therefore, the question by which pathways can systemic inflammation induce brain changes related to cognitive impairment? In this respect the findings of Zenaro and colleagues that neutrophils can promote cognitive decline and AD-like pathology are most interesting. They show in their transgenic animal models that neutrophils can play an important role in the breakdown of the blood-brain barrier (BBB) via the LFA-1 integrin family. Their data that link systemic inflammation directly with BBB damage are in line with clinical observations suggesting that a dysfunctional blood-brain barrier plays a role in the vulnerability of an aging brain to inflammatory mediators.…More
It is well known that patients who have experienced delirium are at risk to develop dementia. An area of controversy is whether delirium is only a marker for a vulnerable brain or if delirium and dementia share common pathogenic mechanism. Most important in this respect is the authors' finding that in their models, transient, early neutrophil blockade has not only direct effect on the improvement of memory but also long-term cognitive benefits six months later.
It can be disputed whether in neutrophils can be found within brain parenchyma in close proximity to Aβ deposits in AD patients as reported by the authors. About 25 years ago we found clusters of cells expressing receptors belonging to the LFA-1 family around amyloid deposits (Rozemuller et al, 1989). However, these cells had the morphological characteristics of microglial cells and not of neutrophils. Furthermore, positive immunolabeling of cells within the brain parenchyma for myeloperoxidase cannot be considered as a specific marker for neutrophils. Finally, the presented data do not show clearly whether the neutrophils passed the vascular cells to reside in the perivascular Virchow-Robin spaces or progressed across the glia limitans into the brain parenchyma.
Although I am not convinced that neutrophils within the brain parenchyma play a role in cognitive impairment and AD pathology, the paper presents strong arguments for a role of neutrophils outside the brain parenchyma. Especially in respect to the question of how systemic inflammation can contribute to cognitive decline in the elderly, and to AD pathology, the authors have identified a most interesting pathway for further experimental and clinical research.
References:
Eikelenboom P, Hoozemans JJ, Veerhuis R, van Exel E, Rozemuller AJ, Van Gool WA. Whether, when and how chronic inflammation increases the risk of developing late-onset Alzheimer's disease. Alzheimers Res Ther. 2012;4(3):15. PubMed.
Fong TG, Davis D, Growdon ME, Albuquerque A, Inouye SK. The interface between delirium and dementia in elderly adults. Lancet Neurol. 2015 Aug;14(8):823-32. Epub 2015 Jun 29 PubMed.
Rozemuller JM, Eikelenboom P, Pals ST, Stam FC. Microglial cells around amyloid plaques in Alzheimer's disease express leucocyte adhesion molecules of the LFA-1 family. Neurosci Lett. 1989 Jul 3;101(3):288-92. PubMed.
Private Health Management
University of Southern California
This study by Zenaro and collaborators focuses on the role of neutrophils in Alzheimer’s disease. These are a type of phagocytotic immune cell that act as first-line defenders to fight inflammation. Studies have observed an increase in neutrophils in AD mouse models and AD patients, but whether or not neutrophils play a role in AD pathogenesis remains unclear (Baik et al., 2014; Kuyumcu et al., 2012).
Zenaro et al. make a number of significant observations. First, the authors show that 4-month old 5xFAD mice develop neurovascular inflammation as suggested by the increased expression of adhesion molecules, such as P-selectin, V-CAM, and I-CAM in brain vessels. This inflammation could lead to blood-brain barrier breakdown, loss of immune privilege, and brain infiltration by neutrophils. Neutrophils are then attracted by and accumulate around Aβ deposits, and secrete cytokines that can damage neurons and the blood-brain barrier. Second, they demonstrate that neutrophil depletion, when administered at early disease stages, ameliorated cognitive performance in 3xTg-AD and 5xFAD mice, which was associated with reduced Aβ load and decreased microgliosis. In addition, blockage of lymphocyte function-associated antigen 1 (LFA-1) integrin, a protein necessary for neutrophil migration and trafficking in the AD brain, reduced pathology and restored memory. Finally, they show that neutrophil depletion could affect synaptic plasticity by restoring presynaptic markers. The presence of neutrophils was also confirmed in human AD brains. …More
Overall, these important data provide direct evidence for the role of neutrophils in AD-related neurodegeneration and cognitive impairment. This provocative study raises a number of interesting questions. Foremost, do these findings have translational potential in regard to human disease? Would targeting neutrophils protect against AD? And what are the possible implications for the development of infections that could also play a role in AD pathogenesis? The study provides potentially new mechanistic insights for the development of novel treatments for AD that focus on controlling the neutrophil response. It will be interesting to see if that pans out in future studies.
References:
Baik SH, Cha MY, Hyun YM, Cho H, Hamza B, Kim DK, Han SH, Choi H, Kim KH, Moon M, Lee J, Kim M, Irimia D, Mook-Jung I. Migration of neutrophils targeting amyloid plaques in Alzheimer's disease mouse model. Neurobiol Aging. 2014 Jun;35(6):1286-92. Epub 2014 Jan 8 PubMed.
Kuyumcu ME, Yesil Y, Oztürk ZA, Kizilarslanoğlu C, Etgül S, Halil M, Ulger Z, Cankurtaran M, Arıoğul S. The evaluation of neutrophil-lymphocyte ratio in Alzheimer's disease. Dement Geriatr Cogn Disord. 2012;34(2):69-74. PubMed.
Make a Comment
To make a comment you must login or register.