Does Huntington’s Disease Begin in the Womb?
Quick Links
Huntington’s disease, like most neurodegenerative conditions, strikes adults. Yet children who carry the infamous causative triple repeat expansion have slightly smaller brains than noncarriers even while they are as functional as their noncarrying siblings, suggesting there could be subtle developmental effects. Supporting this idea, the mutant protein affects embryonic brain development in animal models. In the July 16 Science, researchers led by Sandrine Humbert at INSERM in Grenoble, France, and Alexandra Durr at Sorbonne University in Paris for the first time demonstrate prenatal effects of mutant huntingtin in the human brain. Examining tissue taken from four 13-week-old human fetuses that had inherited the huntingtin gene expansion, they found neural progenitor cells to be abnormal in ways that would likely cause the cells to differentiate too soon, depleting the progenitor pool. This might result in fewer neurons being born.
- In the developing human brain, mutant huntingtin results in the loss of neural progenitor cells.
- This could cause fewer neurons to be born, and explain smaller brain volume in carriers.
- Progenitor cells also were abnormal in their organization, polarity, and endosomal trafficking.
“This is the first direct proof that developmental defects occur in a late-onset neurodegenerative disorder such as Huntington’s disease,” Humbert told Alzforum. The brain appears able to compensate for these abnormalities for decades, however, because carriers remain cognitively healthy until midlife. “By studying how the brain compensates, we may discover new therapeutic targets,” Humbert noted.
“This elegant study extends the previous notion that neurodegenerative disorders are preceded by a long, clinically silent phase of at least several decades that might perhaps even start very early in life,” Marlene Jimenez-Del-Rio of Universidad de Antioquia, Medellin, Colombia, wrote to Alzforum (full comment below). Jimenez-Del-Rio studies autosomal-dominant Alzheimer’s caused by the E280A mutation in presenilin 1, and recently developed a cellular model derived from umbilical cords of newborn mutation carriers (May 2020 news).
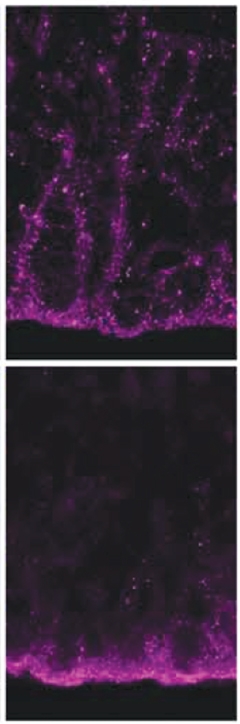
Broken Seal. In developing control brain (top), adhesin protein ZO1 (purple) essentially zip-locks neighboring neuroepithelial cells together; in huntingtin mutation carriers (bottom), ZO1collects at the apical surface of the ventricular zone. [Courtesy of Barnat et al., Science/AAAS.]
Huntington’s disease is caused by an expansion of a CAG repeat in the huntingtin gene. Wild-type huntingtin protein has numerous functions, including helping ferry vesicles along microtubules, regulating cell division and cilia formation, and promoting adhesion of neuroepithelial cells (for review, see Saudou and Humbert, 2016; Lo Sardo et al., 2012).
Mice lacking huntingtin lose forebrain neurons and die in utero (Zeitlin et al., 1995; Reiner et al., 2003). Humbert’s group previously found that mutant huntingtin has similar effects, depleting progenitor cells, derailing migration of newborn neurons, and saddling mice with a thinner cortex (Godin et al., 2010; Barnat et al., 2017; Molina-Calavita et al., 2014).
What about people? Some HD carriers use prenatal genetic testing to avoid passing on this terrible disease to their children. After chorionic villus sampling at 13 weeks revealed the presence of the mutation, four families chose to terminate these pregnancies and to donate the tissue to the authors for research on Huntington’s disease. All four fetuses carried expansions of about 40 repeats, roughly double the normal amount. Humbert and colleagues compared their brains to those from four healthy fetuses from pregnancies terminated for other reasons. Neurodegenerative disease researchers rarely have the opportunity to study such tissue.
In brain slices from the aborted fetuses, first author Monia Barnat examined neural progenitor cells in the ventricular zone. Immunostaining revealed that mutant huntingtin (Htt) appeared stuck at the apical surface of the zone, whereas the wild-type protein was distributed evenly throughout. In addition, mutant Htt misdirected several adhesion proteins, such as ZO1, N-cadherin, and β-catenin, to the apical surface, as well.
Normally these proteins are distributed throughout the ventricular zone, linking progenitor cells together (see image above right). The net effect would likely be to disrupt the close cell contacts and organization of the healthy brain, the authors noted.
At 13 weeks, a still-early stage of human fetal development, progenitor cells are generating the cortical neurons that project to the striatum a few weeks later and, decades later, degenerate in Huntington’s. To do this, their cell bodies migrate between the apical (or ventricular) surface, where they divide, and the basal surface, where they duplicate their DNA. This migration helps maintain the balance between proliferation and differentiation.
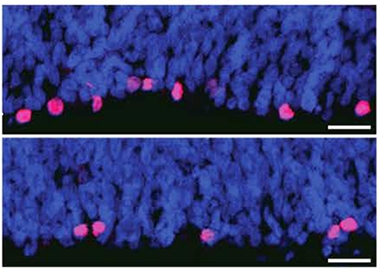
Vanishing Progenitors. The developing brains of HD carriers (bottom) contain fewer mitotic progenitors (pink) than control brains (top). Nuclei in blue. [Courtesy of Barnat et al., Science/AAAS.]
In carriers, apical progenitor cells had more cilia than in controls, implying they spent more time at the apical surface and less time on DNA synthesis. This may favor neuronal differentiation over proliferation (Arai et al., 2011). Moreover, there were fewer dividing cells in carriers than in controls, indicating a smaller progenitor pool (see image at left). Progenitor cells looked abnormal in other ways as well. For example, proteins involved in endosomal transport were mislocalized, implying disruptions in trafficking (see image below).
To explore these hints gleaned from human tissue more deeply in an experimental system, the authors turned to a mouse HD model that carries 111 CAG repeats in the huntingtin gene. Brain slices from 13.5-day-old embryos, the equivalent of 13 weeks of gestation in humans, replicated the changes seen in human tissue. Time-lapse imaging of cortical slices confirmed a slowdown in the cycling of progenitor cells, with less time spent in DNA synthesis. As in human tissue, there were fewer dividing cells, revealing a depletion of progenitors.
Altogether, the findings hint at an HD brain unable to generate a normal number of cortical neurons in certain areas. Previous research has found that children carrying 39 or more CAG repeats have smaller brains on average than noncarriers; the study did not break this down by brain region (Lee et al., 2012). However, other factors, such as a loss of dendritic arborization, could explain these smaller brain volumes as well, Humbert noted.
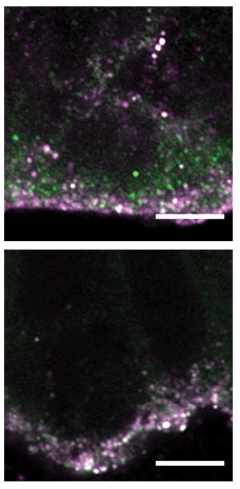
Recycling Failure. In tissue from HD carriers (bottom), recycling endosomes (green) remain stuck at the apical surface of progenitor cells along with huntingtin (purple), in contrast to control tissue (top). [Courtesy of Barnat et al., Science/AAAS.]
Despite these developmental abnormalities, the brain appears able to compensate. Sarah Tabrizi at University College London recently found that young adult HD carriers are cognitively normal. So is their connectivity between the cortex and striatum, although their striata themselves were slightly smaller than those of controls (Scahill et al., 2020). “[T]hey develop normally and function at a high level into adulthood, until the tipping point when gradual neurodegeneration occurs,” Tabrizi wrote to Alzforum (full comment below).
In Alzheimer’s disease as well, some evidence suggests pathobiology could be related in part to developmental effects of the implicated genes, Dennis Selkoe at Brigham and Women’s Hospital, Boston, wrote to Alzforum. Previously, Selkoe and Tracy Young-Pearse at BWH found that amyloid precursor protein is required for neural precursor cells to migrate to the correct position in the cortical plates of embryonic mice (Young-Pearse et al., 2007).
Eric Reiman and colleagues at Banner Alzheimer’s Institute in Phoenix have associated familial APP mutations with hypometabolism and white-matter loss in mice (Gonzalez-Lima et al., 2001; Valla et al., 2008). Children who carry the Paisa presenilin mutation show functional abnormalities on fMRI when performing cognitive tasks, suggesting their brains work less efficiently (Dec 2011 conference news; Jul 2015 news). Some APOE4 carriers have deficits in gray and white matter even as babies, as well as sluggish brain glucose metabolism and subtle problems with their spatial navigation as young adults (Dean et al., 2014; Reiman et al., 2004; Oct 2015 news).
“Evidence of neurodevelopmental effects does raise questions about just how early treatments need to be started in order to be effective,” Nick Fox at UCL wrote to Alzforum. Tabrizi noted that disease-modifying therapies for HD and other neurodegenerative disorders should start as early as possible. Fox agreed, but also noted that people with familial AD mutations are clinically indistinguishable from noncarriers until quite close to symptom onset. “[That] means that there is still great potential for therapies to have a transformative impact even at this stage,” Fox wrote (full comment below).—Madolyn Bowman Rogers
References
News Citations
- Umbilical Cord With Presenilin Mutation Births New Cell Model of Familial AD
- Reeling In Biomarker Data in Young Carriers, API Rocks Staging Boat
- Familial Alzheimer’s Gene Alters Children’s Brains
- Young ApoE4 Carriers Wander Off the ‘Grid’ — Early Predictor of Alzheimer’s?
Paper Citations
- Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016 Mar 2;89(5):910-26. PubMed.
- Lo Sardo V, Zuccato C, Gaudenzi G, Vitali B, Ramos C, Tartari M, Myre MA, Walker JA, Pistocchi A, Conti L, Valenza M, Drung B, Schmidt B, Gusella J, Zeitlin S, Cotelli F, Cattaneo E. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat Neurosci. 2012 May;15(5):713-21. PubMed.
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995 Oct;11(2):155-63. PubMed.
- Reiner A, Dragatsis I, Zeitlin S, Goldowitz D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol Neurobiol. 2003 Dec;28(3):259-76. PubMed.
- Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, Dietrich P, Volvert ML, Guillemot F, Dragatsis I, Bellaiche Y, Saudou F, Nguyen L, Humbert S. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010 Aug 12;67(3):392-406. PubMed.
- Barnat M, Le Friec J, Benstaali C, Humbert S. Huntingtin-Mediated Multipolar-Bipolar Transition of Newborn Cortical Neurons Is Critical for Their Postnatal Neuronal Morphology. Neuron. 2017 Jan 4;93(1):99-114. Epub 2016 Dec 22 PubMed.
- Molina-Calavita M, Barnat M, Elias S, Aparicio E, Piel M, Humbert S. Mutant huntingtin affects cortical progenitor cell division and development of the mouse neocortex. J Neurosci. 2014 Jul 23;34(30):10034-40. PubMed.
- Arai Y, Pulvers JN, Haffner C, Schilling B, Nüsslein I, Calegari F, Huttner WB. Neural stem and progenitor cells shorten S-phase on commitment to neuron production. Nat Commun. 2011 Jan 11;2:154. PubMed.
- Lee JK, Mathews K, Schlaggar B, Perlmutter J, Paulsen JS, Epping E, Burmeister L, Nopoulos P. Measures of growth in children at risk for Huntington disease. Neurology. 2012 Aug 14;79(7):668-74. Epub 2012 Jul 18 PubMed.
- Scahill RI, Zeun P, Osborne-Crowley K, Johnson EB, Gregory S, Parker C, Lowe J, Nair A, O'Callaghan C, Langley C, Papoutsi M, McColgan P, Estevez-Fraga C, Fayer K, Wellington H, Rodrigues FB, Byrne LM, Heselgrave A, Hyare H, Sampaio C, Zetterberg H, Zhang H, Wild EJ, Rees G, Robbins TW, Sahakian BJ, Langbehn D, Tabrizi SJ. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington's disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. 2020 Jun;19(6):502-512. Epub 2020 May 26 PubMed.
- Young-Pearse TL, Bai J, Chang R, Zheng JB, LoTurco JJ, Selkoe DJ. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci. 2007 Dec 26;27(52):14459-69. PubMed.
- Gonzalez-Lima F, Berndt JD, Valla JE, Games D, Reiman EM. Reduced corpus callosum, fornix and hippocampus in PDAPP transgenic mouse model of Alzheimer's disease. Neuroreport. 2001 Aug 8;12(11):2375-9. PubMed.
- Valla J, Gonzalez-Lima F, Reiman EM. FDG autoradiography reveals developmental and pathological effects of mutant amyloid in PDAPP transgenic mice. Int J Dev Neurosci. 2008 May-Jun;26(3-4):253-8. PubMed.
- Dean DC 3rd, Jerskey BA, Chen K, Protas H, Thiyyagura P, Roontiva A, O'Muircheartaigh J, Dirks H, Waskiewicz N, Lehman K, Siniard AL, Turk MN, Hua X, Madsen SK, Thompson PM, Fleisher AS, Huentelman MJ, Deoni SC, Reiman EM. Brain differences in infants at differential genetic risk for late-onset Alzheimer disease: a cross-sectional imaging study. JAMA Neurol. 2014 Jan;71(1):11-22. PubMed.
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004 Jan 6;101(1):284-9. PubMed.
Further Reading
Primary Papers
- Barnat M, Capizzi M, Aparicio E, Boluda S, Wennagel D, Kacher R, Kassem R, Lenoir S, Agasse F, Braz BY, Liu JP, Ighil J, Tessier A, Zeitlin SO, Duyckaerts C, Dommergues M, Durr A, Humbert S. Huntington's disease alters human neurodevelopment. Science. 2020 Aug 14;369(6505):787-793. Epub 2020 Jul 16 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
These data are very interesting. There has been debate in the Huntington’s disease field regarding the existence of a neurodevelopmental deficit. Evidence is accruing that this may be the case based on differentiating HD IPSc systems, mouse development, and now these studies of early human development.
We recently found that HD gene carriers ~24 years before predicted disease onset had essentially completely normal brains, including normal cortico-striatal connectivity on advanced neuroimaging, apart from a slightly smaller striatum, which we hypothesized resulted in selective vulnerability of the striatum to subsequent neurodegeneration in HD (Scahill et al., 2020).
Importantly, our HD gene carriers performed as well as matched controls on a range of stringent cognitive and motor assessments. Our unpublished functional imaging data in this cohort suggests compensation occurs in these HD gene carriers.
Therefore, despite these neurodevelopmental deficits, they develop normally and function at a high level into adulthood, but there is a tipping point when gradual neurodegeneration occurs. This all suggests that we need to treat as early as possible with disease-modifying therapies to enable us to delay or prevent symptom onset, and it means that there is still great potential for therapies to potentially prevent the neurodegeneration occurring if we treat early enough. We need to understand more about the very earliest manifestations of neurodegeneration and then intervene at the optimal stage.
References:
Scahill RI, Zeun P, Osborne-Crowley K, Johnson EB, Gregory S, Parker C, Lowe J, Nair A, O'Callaghan C, Langley C, Papoutsi M, McColgan P, Estevez-Fraga C, Fayer K, Wellington H, Rodrigues FB, Byrne LM, Heselgrave A, Hyare H, Sampaio C, Zetterberg H, Zhang H, Wild EJ, Rees G, Robbins TW, Sahakian BJ, Langbehn D, Tabrizi SJ. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington's disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. 2020 Jun;19(6):502-512. Epub 2020 May 26 PubMed.
Dementia Reserch Center
This is an interesting paper. In other single-gene neurodegenerative diseases, from prion to Alzheimer’s, there have been suggestions of potential neurodevelopmental influences, but this study takes an interesting approach to addressing this through the assessment of fetal tissue.
Our understanding of familial AD has changed over recent decades with very extensive evidence to support a long preclinical period. That period extends over 20 years or so, with progressive accumulation of molecular pathology being followed by neurodegenerative, functional, and cognitive changes—all of which are insidious in onset and relentlessly progressive before diagnosis.
Evidence of neurodevelopmental effects does raise questions about just how early treatments need to be started in order to be effective, or, as the authors put it, to be "sufficient to forestall symptom progression.” However, in my view, the fact that carriers of mutations for Alzheimer’s disease function to a high level into adulthood, and in fact are effectively indistinguishable from their noncarrier siblings until quite close to symptom onset or clinical diagnosis, means that there is still great potential for therapies to have a transformative impact even at this stage. There is the potential for disease modification to be effective "secondary prevention" and to maintain “normal” cognitive function. We need those therapies.
University of Antioquia
This elegant study extends the previous notion that neurodegenerative disorders are preceded by a long, clinically silent phase of at least several decades that might perhaps even start very early in life. Unsurprisingly, several aspects of mHTT mirror familial Alzheimer’s disease bearing PSEN1 mutations, e.g., PSEN1 E280A.
For example, both disorders present at least five main common features: (i) accumulation of intracellular protein HTT and sAPPβ fragments, (ii) impairment of endosome secretion and recycling in HD and endosome intoxication by sAPPβ fragments, (iii) alterations in neuronal progenitor proliferation, (iv) mHTT and APP fragments (including Aβ42) alter early stages of brain development in human HD and AD; and (v) children show probably compensatory functional mechanisms which mask apparent abnormality in those individuals bearing mHTT and PSEN1 E280A.
Therefore, it is reasonable to think that treatment for both HD and FAD should start very early in life. I support the view that a developmental component in both progressive neurodegenerative disorders is now unquestionable. We anticipate that this sort of study might stimulate new therapeutic design for HD and FAD patients.
Make a Comment
To make a comment you must login or register.